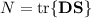4.4 The General Options Menu
After you specified all data concerning the molecule you want to examine, you are on your
way to the last of the four main menus. Before reaching it, you will perhaps get a message
like the following:
DO YOU WANT TO DELETE DATA GROUPS LIKE
$energy
$grad
$hessian
$hessian (projected)
$last energy change
$maximum norm of internal gradient
$dipgrad
$vibrational normal modes
$vibrational spectrum
$cartesianforce interspace
LEFT OVER FROM PREVIOUS CALCULATIONS ? DEFAULT(n)
define has scanned your input file for this session and found some data groups which
might have become obsolete. If they are still acceptable depends on the changes you made
during your present define session. They are obviously incorrect if you changed the
molecule under consideration; but any change in the basis sets or the occupation numbers
will make them dangerous, too, because you might not know some day if they really refer
to the basis set which is defined in this control file. As a rough guide, delete them
whenever you have made changes in one of the first three main menus during your
define session.
After that you will reach the last main menu of define which helps you to control the
actions of all TURBOMOLE programs. The meanings of the various options are explained in
more detail in the description of the individual programs, therefore only a short
explanation will be given here.
Now have a look at the menu:
GENERAL MENU : SELECT YOUR TOPIC
scf : SELECT NON-DEFAULT SCF PARAMETER
mp2 : OPTIONS AND DATA GROUPS FOR rimp2 and mpgrad
pnocc : OPTIONS AND DATA GROUPS FOR pnoccsd
cc : OPTIONS AND DATA GROUPS FOR ricc2
ex : EXCITED STATE AND RESPONSE OPTIONS
prop : SELECT TOOLS FOR SCF-ORBITAL ANALYSIS
drv : SELECT NON-DEFAULT INPUT PARAMETER FOR EVALUATION
OF ANALYTICAL ENERGY DERIVATIVES
(GRADIENTS, FORCE CONSTANTS)
rex : SELECT OPTIONS FOR GEOMETRY UPDATES USING RELAX
stp : SELECT NON-DEFAULT STRUCTURE OPTIMIZATION PARAMETER
e : DEFINE EXTERNAL ELECTROSTATIC FIELD
dft : DFT Parameters
ri : RI Parameters
rijk : RI-JK-HF Parameters
rirpa : RIRPA Parameters
senex : seminumeric exchange parameters
hybno : hybrid Noga/Diag parameters
dsp : DFT dispersion correction
trunc : USE TRUNCATED AUXBASIS DURING ITERATIONS
marij : MULTIPOLE ACCELERATED RI-J
dis : DISPLAY MOLECULAR GEOMETRY
list : LIST OF CONTROL FILE
& : GO BACK TO OCCUPATION/ORBITAL ASSIGNMENT MENU
* or q : END OF DEFINE SESSION
This menu serves very different purposes. The next subsection deals with commands
required to activate and/or specify specific methods of calculation. The subsequent
subsection describes commands used to select non-default options. Standard SCF
calculations do not require special action, just leave the menu. The final subsection
describes the settings for property calculations.
4.4.1 Important commands
DFT calculations
Command dft leads you to the menu:
STATUS OF DFT_OPTIONS:
DFT is NOT used
functional b-p
gridsize m3
ENTER DFT-OPTION TO BE MODIFIED
func: TO CHANGE TYPE OF FUNCTIONAL
grid: TO CHANGE GRIDSIZE
on: TO SWITCH ON DFT
Just <ENTER>, q or ’*’ terminate this menu.
To activate DFT input on and then specify the grid for the quadrature of
exchange-correlation terms. TURBOMOLE offers grids 1 (coarse) to 7 (finest), and the
multiple grids m3 to m5 [4]. The latter employ a coarser grid during SCF iterations, and
grid 3 to grid 5 in the final SCF iteration and the gradient evaluation. Default is grid m3,
for clusters with more than 50 atoms use m4.
The functionals supported are obtained with the command func:
SURVEY OF AVAILABLE EXCHANGE-CORRELATION ENERGY FUNCTIONALS
FUNCTIONAL | TYPE | EXCHANGE | CORRELATION | REFERENCES
---------------------------------------------------------------------
slater-dirac- | LDA | S | | 1,2
exchange | | | |
s-vwn | LDA | S | VWN(V) | 1-3
vwn | LDA | | VWN(V) | 3
s-vwn_Gaussian | LDA | S | VWN(III) | 1-3
pwlda | LDA | S | PW | 1,2,4
becke-exchange | GGA | S+B88 | | 1,2,5
b-lyp | GGA | S+B88 | LYP | 1,2,6
b-vwn | GGA | S+B88 | VWN(V) | 1-3,5
lyp | GGA | | LYP | 6
b-p | GGA | S+B88 | VWN(V)+P86 | 1-3,5,7
pbe | GGA | S+PBE(X) | PW+PBE(C) | 1,2,4,8
tpss | MGGA | S+TPSS(X) | PW+TPSS(C) | 1,2,4,14
bh-lyp | HYB | 0.5(S+B88) | LYP | 1,2,5,6,9
| | +0.5HF | |
b3-lyp | HYB | 0.8S+0.72B88 | 0.19VWN(V) | 1-3,5,6,10
| | +0.2HF | +0.81LYP |
b3-lyp_Gaussian | HYB | 0.8S+0.72B88 | 0.19VWN(III) | 1-3,5,6,10
| | +0.2HF | +0.81LYP |
pbe0 | HYB | 0.75(S+PBE(X)) | PW+PBE(C) | 1,2,4,8,11
| | +0.25HF | |
tpssh | HYB | 0.9(S+TPSS(X)) | PW+TPSS(C) | 1,2,4,14,15
| | +0.1HF | |
lhf | EXX | EXX | | 12,13
b97-d | GGA | B97 refit | B97 refit | 16
b2-plyp | DHYB |0.47(SB88)+0.53HF|0.73LYP+0.27PT2| 17
Default is b-p, i.e. B-P86, which is probably best for the whole of Chemistry [31]. For
main group compounds we recommend b3-lyp; note that GAUSSIAN uses partly different
implementations [31].
The programs dscf and grad are used to carry out conventional DFT treatments, i.e. J
and K are evaluated without approximations.
RI-J calculations
For non-hybrid functionals we strongly recommend the RI-J procedure, which speeds up
calculations by a factor 10 at least (as compared to conventional treatments) without
sacrificing accuracy. Command ri gives:
STATUS OF RI-OPTIONS:
RI IS NOT USED
Memory for RI: 200 MB
Filename for auxbasis: auxbasis
ENTER RI-OPTION TO BE MODIFIED
m: CHANGE MEMORY FOR RI
f: CHANGE FILENAME
jbas: ASSIGN AUXILIARY RI-J BASIS SETS
on: TO SWITCH ON RI
Use <ENTER>, q, end, or * to leave this menu
Activate RI-J with on, and choose with m the memory you can dedicate to store
three-center integrals (Keyword: $ricore), default is 200MB. The more memory, the
faster the calculation.
A rough guide: put $ricore to about 2∕3 of the memory of the computer. Use OS specific
commands (top on most UNIX systems), during an ridft run to find the actual
memory usage and then adjust $ricore, the keyword in control specifying
memory.
If the option jbas is selected, define enters a submenu which allows the assignment of
auxiliary basis sets (for an explanation of the menu items see Section 4.2). Where
available, the program will select by default the auxiliary basis sets optimized for the
orbital basis used. Please note that treatment of systems with diffuse wavefunctions may
also require an extension of the auxiliary basis. For this cases enlarge the sets of s- and
p-functions with diffuse functions.
The RI-J option is only supported by programs ridft and rdgrad, if you use jobex to
optimize molecular geometry, put: nohup jobex -ri ...
MARI-J option
RI-J calculations can be done even more efficiently with the Multipole Accelerated RI-J
(MARI-J) option, especially for larger molecules where almost linear scaling is
achieved [32].
Parameters:
1) precision parameter: 1.00E-06
2) maximum multipole l-moment: 10
3) maximum number of bins: 8
4) minimum separation of bins: 0.00
5) maximum allowed extension: 20.00
6) threshold for multipole neglect: 1.00E-18
Enter the number to change a value or <return> to accept all.
Just rely on the defaults.
Multiple auxiliary basis sets
With the command trunc you can switch on this option. Effect: a reduced auxiliary (or
fitting) basis to represent the electron density is employed during SCF iterations,
the final SCF iteration and the gradient are computed with the full auxiliary
basis.
truncated RI ALREADY SWITCHED ON
DO YOU WANT TO SWITCH OFF truncation ? (default=no)
Note: trunc is presently not compatible with marij!
RI in SCF calculations
Considerable savings in CPU times are achieved with the RI technique for both Coulomb
J and exchange K terms in SCF calculations, the RI-JK method [33], provided
large basis sets are employed, e.g. TZVPP, cc-pVTZ, or cc-pVQZ. With rijk you
get:
STATUS OF RI-OPTIONS:
RI IS NOT USED
Memory for RI: 200 MB
Filename for auxbasis: auxbasis
ENTER RI-OPTION TO BE MODIFIED
m: CHANGE MEMORY FOR RI
f: CHANGE FILENAME
jkbas: ASSIGN AUXILIARY RI-JK BASIS SETS
on: TO SWITCH ON RI
Use <ENTER>, q, end, or * to leave this menu
For an explanation of the menu items see Section 4.4.1. RI-JK calculations can be
carried out with the program ridft.
Optimization to minima and transition structures using STATPT
Structure optimizations can be carried out by the program statpt. For minimizations no
additional keywords are required. The default values are assumed, which work in most of
the cases. Structure optimization is performed in internal coordinates if they
have been set. Otherwise, Cartesian coordinates are used. One can switch the
optimization in internal coordinates on or off, respectively in internal redundant or
cartesian coordinates. For transition structure optimizations the index of transition
vector has to be set to an integer value > 0 (0 means structure minimization).
The value of the index specifies transition vector to follow during the saddle
point search. Note, that Hessian eigenpairs are stored in ascending order of the
eigenvalues, i.e. the eigenpair with the smallest eigenvector has the index 1.
The command stp gives:
------------------------------------------------------------------------
CONVERGENCE CRITERIA:
thre 1.000000E-06 thre : threshold for ENERGY CHANGE
thrd 1.000000E-03 thrd : threshold for MAX. DISPL. ELEMENT
thrg 1.000000E-03 thrg : threshold for MAX. GRAD. ELEMENT
rmsd 5.000000E-04 rmsd : threshold for RMS OF DISPL.
rmsg 5.000000E-04 rmsg : threshold for RMS OF GRAD.
defl : set default values.
------------------------------------------------------------------------
OPTIMIZATION refers to
int off int: INTERNAL coordinates
rdn off rdn: REDUNDANT INTERNAL coordinates
crt on crt: CARTESIAN coordinates
NOTE : options int and crt exclude each other
ENTER STATPT-OPTIONS TO BE MODIFIED
itvc 0 itvc : change INDEX OF TRANSITION VECTOR
updte bfgs updte: change method of HESSIAN UPDATE
hsfrq 0 hsfrq: frequency of HESSIAN CALCULATION
kptm 0 kptm : FREEZING transition vector INDEX
hdiag 5.000000E-01 hdiag: change DIAGONAL HESSIAN ELEMENTS
rmax 3.000000E-01 rmax : change MAX. TRUST RADIUS
rmin 1.000000E-04 rmin : change MIN. TRUST RADIUS
trad 3.000000E-01 trad : change TRUST RADIUS
------------------------------------------------------------------------
Just <ENTER>, q or ’*’ terminate this menu.
Excited states, frequency-dependent properties, and stability analysis
Excited state calculations with RPA or CIS (based on HF-SCF) and TDDFT
procedures as well as stability analyses (SCF or DFT) are carried out by the program
escf.
You will need a well converged HF-SCF or DFT calculation that were converged to at
least $scfconv=7, see Section 4.4.2.
Details of calculations are specified with the command ex:
MAIN MENU FOR RESPONSE CALCULATIONS
OPTION | STATUS | DESCRIPTION
-------------------------------------------------------------------
rpas | off | RPA SINGLET EXCITATIONS (TDHF OR TDDFT)
ciss | off | TDA SINGLET EXCITATIONS (CI SINGLES)
rpat | off | RPA TRIPLET EXCITATIONS (TDHF OR TDDFT)
cist | off | TDA TRIPLET EXCITATIONS (CI SINGLES)
polly | off | STATIC POLARIZABILITY
dynpol | off | DYNAMIC POLARIZABILITY
single | off | SINGLET STABILITY ANALYSIS
triple | off | TRIPLET STABILITY ANALYSIS
nonrel | off | NON-REAL STABILITY ANALYSIS
ENTER <OPTION> TO SWITCH ON/OFF OPTION, * OR q TO QUIT
If you have selected an option, e.g. rpas, and quit this menu, you will get another
menu:
SELECT IRREP AND NUMBER OF STATES
ENTER ? FOR HELP, * OR Q TO QUIT, & TO GO BACK
This should be self-evident.
MP2, MP2-F12, RI-MP2, and PNO-MP2
We recommend to use MP2 for standard applications together with the RI technique using
the ricc2 program. The mpgrad program is supplied for special benchmark application
where the RI approximation needs to be avoided. The pnoccsd program is meant only for
very large (> 100 atoms and > 3000 basis functions) MP2 and MP2-F12 single point
energy calculations. This is more efficient and supports the frozen core option in the
gradient calculation.
The entry mp2 leads to a submenu which allows to set some keywords for MP2 and
RI-MP2 calculations, e.g. defining frozen orbitals, maximum memory usage, or assign
auxiliary basis sets for RI-MP2 calculations, etc. If you want to use ricc2, you have to use
the entry cc and the submenu ricc2 in order to assign MP2 as wavefunction model. For
the pnoccsd program you have to use the entry pnocc and the submenu pnoccsd to assign
the wavefunction model.
Conventional MP2 calculations with mpgrad require a number of additional settings for
which it is recommended to invoke the interactive tool mp2prep. For geometry
optimizations with jobex use nohup jobex -level mp2 -ri ...
CC2 and CCSD calculations
The entry cc leads to a submenu which allows to set a number of keywords essential for
calculations with the programs ricc2 and ccsdf12. In particular it allows the assignment
of the wavefunction method(s), selection of auxiliary basis sets, the specification of frozen
orbitals, and the definition of a scratch directory and of the maximum core memory
usage.
The ricc2 program must be used for excitation energies and response properties with
second-order methods (MP2, CIS(D), ADC(2), CC2, etc. and their spin-scaled variants),
while the ccsdf12 program must be used for third- and higher-order methods (MP3,
CCSD, CCSD(T), etc.).
2nd analytical derivatives for SCF and DFT
The program aoforce computes force constants and IR and Raman Spectra on SCF and
DFT level. Analytical second derivative calculations can directly be started from
converged SCF or DFT calculations. Note, that the basis is restricted to d-functions, and
ROHF as well as broken occupation numbers are not allowed. For better efficiency, in
case of larger systems, use the keyword $maxcor as described in Chapter 14 to
reduce computational cost. RI will be used if the RI option for DFT has been
specified.
4.4.2 Special adjustments
Adjustments described by the following menus are often better done directly in the
control file; have a look at the keywords in Chapter 20. For common calculations just
start with the defaults, and change keywords directly in control if you encounter
problems with your calculation.
SCF options
ENTER SCF-OPTION TO BE MODIFIED
conv : ACCURACY OF SCF-ENERGY $scfconv
thi : INTEGRAL STORAGE CRITERIA $thize $thime
ints : INTEGRAL STORAGE ALLOCATION $scfintunit
iter : MAXIMUM NUMBER OF ITERATIONS $scfiterlimit
diis : DIIS CONVERGENCE ACCELERATION $scfdiis
damp : OPTIONS FOR DAMPING $scfdamp
shift: SHIFTING OF ORBITALS $scforbitalshift
order: ORDERING OF ORBITALS $scforbitalorder
fermi: THERMAL SMEARING OF OCC. NUMBERS $fermi
By the command $fermi you can switch on smearing of occupation numbers, and thus
automatically optimize occupations and spin.
Menu drv
The most important of the derivative menus is the first one which tells the programs which
derivatives to calculate. This is only necessary for special purposes and you should better
not change default options.
------------------------------------------------------------------------
derivative data groups ’$drvopt, $drvtol’
------------------------------------------------------------------------
option | status | description :
------------------------------------------------------------------------
crt | T | CARTESIAN 1st derivatives
sec | T | CARTESIAN 2nd derivatives
bas | F | energy derivatives with respect to
| | BASIS SET exponents/scaling factors/
| | contraction coefficients
glb | F | energy derivative with respect to
| | a GLOBAL scaling factor
dip | T | cartesian 1st derivatives of DIPOLE MOMENT
pol | T | nuclear contribution to POLARIZABILITY
fa | F | SPECTROSCOPIC ANALYSIS only
tol 0.100D-06 derivative integral cutoff
------------------------------------------------------------------------
use <opt> for enabling, -<opt> for disabling of logical switches
<&> will bring you back to GENERAL MENU without more changes
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
The handling of these options is very simple. With the exception of tol, all are logical
switches which are either true (or on, active) or false (or off, inactive). You can switch
between the two states if you enter, for example, crt (to switch calculation of Cartesian
first derivatives on) or -crt (to switch it off). The options crt, sec and bas
should provide no problems. glb refers to a global scaling factor for all basis set
exponents. Imagine that you would like to replace your basis set, which contains basis
functions
by another basis set which contains basis functions
where α is the same for all primitive basis functions χμ. With command glb you are able
to calculate analytical derivatives of the total energy with respect to α and can thus easily
determine the optimum α.
dip enables you to calculate the first derivatives of the electric dipole moment with
respect to nuclear displacements which gives you infrared intensities. pol allows
you to calculate the contribution of the nuclear rearrangement on the electric
polarizability. fa finally performs only a frequency analysis which means that
aoforce will read the force constant matrix ($hessian or $hessian (projected)),
diagonalize it and give you the frequencies and normal modes. tol is not a logical
switch as the other options in this menu, but a cutoff threshold for the derivative
integrals, i.e. integrals below this threshold will be neglected in the derivative
calculations.
Entering * will bring you to the second derivative submenu.
Debug Options for the Derivative Programs
The following menu deals only with some debug options for grad. Use them with caution,
each of them can produce lots of useless output:
------------------------------------------------------------------------
derivative debug options ’$drvdebug’
------------------------------------------------------------------------
option |status| description :
------------------------------------------------------------------------
disp1e | F | display 1e contributions to desired derivatives
only1e | F | calculate 1e contributions to desired derivatives only
debug1e | F | display 1e shell contributions to desired derivatives
| | (WARNING : this produces large outputs!)
debug2e | F | display 2e shell contributions to desired derivatives
| | (WARNING : this produces VERY large outputs!)
debugvib| F | debug switch for vibrational analysis (force only)
notrans | F | disable transfer relations (gradient only!)
novirial| F | disable virial scaling invariance in basis set
| | optimizations (gradient only)
------------------------------------------------------------------------
use <opt> for enabling, -<opt> for disabling option <opt>
<&> will bring you back to GENERAL MENU without more changes
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
As there is no need to use these options normally and the menu text is self-explaining, no
further description will be given. Note that all options are logical switches and may be
enabled and disabled the same way as shown for the last menu. Entering * will bring you
to the last derivative submenu.
4.4.3 Relax Options
Program relax has a huge variety of options to control its actions which in program
define are grouped together in eight consecutive menus. These are only briefly described
in the following sections; for a more detailed discussion of the underlying algorithms refer
to the documentation of program relax (see Section 5.3). Only experts should try to
change default settings.
Optimization Methods
The first of the relax subgenus deals with the type of optimization to be performed:
------------------------------------------------------------------------
optimization options for RELAX
------------------------------------------------------------------------
option | status | description : optimization refers to
------------------------------------------------------------------------
int | F | INTERNAL coordinates
crt | F | CARTESIAN coordinates
bas | F | BASIS SET exponents/scale factors
glb | F | GLOBAL scaling factor
------------------------------------------------------------------------
use <opt> for enabling, -<opt> for disabling option <opt>
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
You can choose between a geometry optimization in the space of internal coordinates (in
this case you will need definitions of internal coordinates, of course) or in the space of
Cartesian coordinates (these possibilities are mutually exclusive, of course). Furthermore
optimizations of basis set parameters (exponents, contraction coefficients and scaling
factors) or of a global scaling factor is possible (these options are also exclusive, but can be
performed simultaneous to a geometry optimization). For the geometry optimization you
should normally use internal coordinates as they provide better convergence characteristics
in most cases.
Coordinate Updates
The next submenu deals with the way relax updates the old coordinates. You may choose
a maximum change for the coordinates or you can allow coordinate updates by means of
extrapolation:
------------------------------------------------------------------------
coordinate update options for RELAX
------------------------------------------------------------------------
dqmax <real> : coordinates are allowed to change by at most
<real> (DEFAULT : 0.3000 ) a.u.
polish : perform an interpolation or extrapolation of
coordinates (DEFAULT :y)
-polish : disable inter/extrapolation
------------------------------------------------------------------------
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
These options result in better convergence of your optimization in most cases.
Interconversion Between Internal and Cartesian Coordinates
The interconversion between internal and Cartesian coordinates is not possible directly (in
this direction). Instead it is performed iteratively. The following options control this
conversion:
--------------------------------------------------------------------
interconversion options for RELAX
--------------------------------------------------------------------
option | description
--------------------------------------------------------------------
on | switch on interconversion (DEFAULT: off)
qconv <r> | set convergence threshold for interconversion
| of coordinates to <r>. DEFAULT : <r> = .1000E-09
iter <i> | allow at most <i> iterations for interconversion
| of coordinates. DEFAULT : <i> = 25
crtint | transform cartesian into internal coordinates (DEFAULT=n)
intcrt | transform internal into cartesian coordinates (DEFAULT=n)
grdint | transform cartesian into internal gradients (DEFAULT=n)
hssint | transform cartesian into internal hessian (DEFAULT=n)
--------------------------------------------------------------------
use -<opt> for disabling any interconversion option
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
The options qconv and iter are used in each normal relax run to determine
the characteristics of the back-transformation of coordinates into the internal
space. With the other options and interconversion switched on, you can force
relax to perform only the specified coordinate transformation and write the
transformed coordinates to file control. To achieve this, enter on to switch to the
transformation-only mode, and one of the last four options, e.g. crtint, to specify the
desired transformation.
Updating the Hessian
relax provides a variety of methods to generate an updated Hessian every cycle. This
includes the well known methods such as BFGS, DFP, or MS update methods as well as
some less common procedures:
--------------------------------------------------------------
OPTIONS FOR UPDATING THE HESSIAN
--------------------------------------------------------------
option | status | description
--------------------------------------------------------------
none | F | NO UPDATE (STEEPEST DESCENT)
bfgs | F | BROYDEN-FLETCHER-GOLDFARB-SHANNO UPDATE
dfp | F | DAVIDON-FLETCHER-POWELL UPDATE
bfgs-dfp | F | COMBINED (BFGS+DFP) UPDATE
ms | F | MURTAGH-SARGENT UPDATE
schlegel | F | SCHLEGEL UPDATE
diagup | F | DIAGONAL UPDATE (AHLRICHS/EHRIG)
multidim | F | RANK > 2 BFGS-TYPE UPDATE
ahlrichs | T | MACRO : AHLRICHS UPDATE (DEFAULT)
--------------------------------------------------------------
USE <opt> FOR ENABLING OPTION <opt> AND THUS DISABLING
ALL OTHER OPTIONS.
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
We recommend to use the default method ahlrichs which provides excellent convergency
in most cases.
General Boundary Conditions for Update
The force constant matrix will only be updated if least mingeo cycles exist. The maximum
number of cycles used for the update is specified by the parameter maxgeo. Normally the
default values provided by define need not be changed.
DEFINE BOUNDARY CONDITIONS FOR UPDATE
--------------------------------------------------------------
mingeo <i> | START UPDATE IF THERE ARE AT LEAST <i> CYCLES
| DEFAULT : min 3
maxgeo <i> | USE LAST <i> CYCLES FOR UPDATE, DEFAULT : max 4
--------------------------------------------------------------
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
Special Boundary Conditions for Ahlrichs and Pulay Updates
For the default update method ahlrichs some additional control parameters are available
which can be defined in this menu:
DEFINE BOUNDARY CONDITIONS FOR AHLRICHS OR PULAY UPDATE
--------------------------------------------------------------
option | description
--------------------------------------------------------------
modus <i> | DEFINE MODUS FOR GDIIS PROCEDURE : MINIMIZE
| <dq|dq> IF <i> = 0
| <g|dq> IF <i> = 1
| <g|g> IF <i> = 2
| <dE> IF <i> = 3
| DEFAULT : <i> = 1
fail <r> | IGNORE GDIIS IF <g|dq> /| <g|dq> | IS
| LARGER THAN -<r>. DEFAULT : <r> = 0.1
--------------------------------------------------------------
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
For detailed description consult Section 5.3.
--------------------------------------------------------------
OPTIONS FOR MANIPULATING THE HESSIAN
--------------------------------------------------------------
option | description
--------------------------------------------------------------
diagonal | RESTRICT UPDATE TO DIAGONAL-ELEMENTS IF
| METHOD IS BFGS,DFP OR MS. DEFAULT=n
offreset | DISCARD OFF-DIAGONAL ELEMENTS. DEFAULT=n
offdamp <r> | DAMP OFF-DIAGONAL ELEMENTS BY 1/(1+<r>) DEFAULT= 1.000
damp <real> | DAMP UPDATE BY 1/(1+<real>), DEFAULT= .0000E+00
scale <real> | SCALE INPUT HESSIAN BY <real>, DEFAULT= 1.000
allow <real> | SCALE INPUT HESSIAN BY <real>/|DE| IF |DE|,
| THE OBSERVED ABSOLUTE CHANGE IN ENERGY, IS
| OBEYING THE CONDITION |DE| > <real> > 0.
| DEFAULT : NO SCALING
min <real> | DO NOT ALLOW EIGENVALUES OF HESSIAN TO DROP
| BELOW <real>. DEFAULT= .1000E-02
reset <real> | USE <real> AS A RESET VALUE FOR TOO SMALL
| EIGENVALUES (CP. min). DEFAULT= .1000E-02
max <real> | DO NOT ALLOW EIGENVALUES OF HESSIAN TO BECOME
| LARGER THAN <real>. DEFAULT= 1000.
--------------------------------------------------------------
WITH THE EXCEPTION OF min,reset AND max, ALL OPTIONS MAY BE
DISABLED BY ENTERING -<opt>
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
Initialization of the Hessian
Finally there are some options to control the choice of the initial Hessian during your
geometry optimization:
--------------------------------------------------------------
FORCE CONSTANTS INITIALIZATION OPTIONS FOR RELAX
--------------------------------------------------------------
OPTION | DESCRIPTION
--------------------------------------------------------------
off | switch off initialization (DEFAULT: on)
cart | use analytical cartesian hessian provided by a
| 2nd derivatives calculation. DEFAULT(n)
diag | use diagonal matrix with diagonal elements set
| individually within data groups $intdef or
| $basis or $global. DEFAULT(n)
unit <r> | use multiple of the unit matrix ( H = <r>*E ).
| DEFAULT(n) - DEFAULT <r> = 1.000
--------------------------------------------------------------
NOTE THAT THESE OPTIONS ARE MUTUALLY EXCLUSIVE
<RETURN> OR * OR q(uit) WILL TERMINATE THIS MENU
Option off will be used if you have already a good Hessian from a previous calculation
which may be used. cart describes an even better state where you have a Hessian from a
calculation of the second derivatives available (aoforce). The other two options describe
real procedures for initialization of the Hessian. Default values: stretches (0.5), angles
(0.2).
4.4.4 Definition of External Electrostatic Fields
This submenu allows you to calculate first and second numerical derivatives of the
energy with respect to an external electric field. The first three options should be
clear; 1st and 2nd are logical switches which are turned on and off the usual way
(1st or -1st) and delta is the increment for the numerical differentiation, that
is, the finite value of the external field, which replaces the (ideally) differential
field:
--------------------------------------------------------------------
electrostatic field definition menu
--------------------------------------------------------------------
option | status | description
--------------------------------------------------------------------
1st | F | numerical 1st derivative dE/dField
2nd | F | numerical 2nd derivative d2E/dField2
delta <real> | | increment for numerical differentiation
| | DEFAULT = .5000E-02
geofield | F | geometry optimization with external field
man | F | explicit definition of electrostatic field(s)
--------------------------------------------------------------------
geofield gives the possibility to perform a whole geometry optimization under the
influence of a finite external field and thus to obtain the (distorted) minimum geometry in
this field. To do this, an external electrostatic field must be defined explicitly which can be
done using command man. Note that geofield must also be switched on if any properties
are to be evaluated in the presence of an electric field. The most prominent example is the
calculation of hyperpolarizabilies.
Take Care, due to some inconsistencies in define it is always necessary to switch on the
field calculations manually. Therefore edit the control file after having finished your
define session and enter on after the entries of fields and geofield.
4.4.5 Properties
The program moloch used for this purpose is currently being revamped, and will then be
much simpler to use. The subsequent description for an older version may not work in all
cases—sorry for that.
If you enter prop in the general menu, define first will check whether the data group
$properties does already exist in your control file or in a file referenced therein. If this
is not the case you will be asked to specify the file on which $properties shall be
written:
data group $properties has not yet been specified
FOR INITIALIZING <moloch> KEYWORDS ENTER
[return] : WRITE TO CONTROL FILE control (DEFAULT), OR
filename : WRITE TO ANOTHER FILE
Afterwards you will get the following submenu which allows you to control all possible
actions of program moloch:
switch on one or more of the following options <i>
<i> = 1,..., 9
for switching off option <i>, specify -<i>
( 1) trace off
( 2) moments off
( 3) potential off
( 4) cowan-griffin off
( 5) localization off
( 6) population analyses off
( 7) plot off
( 8) firstorder off
selecting an already active option indicates that
suboptions shall be modified
* or q(uit) = quit | for help, type help <integer>
All options in this menu are selected by entering their number as indicated in the first
column. For example, to switch on option trace enter 1. The flag off will then change to
active. To switch off an option enter its negative number, e.g. -1 for trace. Most of the
options require additional input and will therefore lead you to further submenus. These are
briefly described below.
Option trace
trace will calculate the trace of density times overlap matrix:
If the orbitals are orthonormal, N should yield the total number of electrons in your
molecule. If this is not true, your MO-vector will most probably be erroneous. For
example, the vector might belong to another geometry or basis set. As this is a very
sensitive test for errors like these and the calculation requires almost no time, you should
always switch on this option.
Option moments
This option leads you to the following submenu:
add/change options for data group $moments
option | status | description
------------------|--------|-------------------------------
point <x> <y> <z> | T | reference point = (x,y,z)
atom <i> | F | reference point = atom no. <i>
0th | T | compute 0th moment
1st | F | compute 1st moment
2nd | F | compute 2nd moment
3rd | F | compute 3rd moment
------------------|--------|-------------------------------
-<moment> : skip computation of <moment>
* or q(uit) : terminate input
This menu serves to specify the electrostatic moments to be calculated (0th=charge,
1st=dipole moment, 2nd=quadrupole moment, 3rd=octuple moment). The reference
point is the origin of the coordinate system used in the calculation. The value of any
calculated moment will be independent of this reference point, if all lower moments are
zero. The default for the reference point is the origin, i.e. the coordinate system used for
the calculation of the moments will be the same as the one in which the atomic
coordinates are specified. The reference point may be changed by typing point
with the three new coordinates appended. Alternatively you may choose the
coordinates of one of the atoms as reference point by entering atom and the atom
index.
Option potential
This option collects all possible quantities related to the electrostatic field created by the
molecular charge distribution. This includes the following suboptions:
list of suboptions :
pot - electrostatic potential
fld - electrostatic field
fldgrd - electrostatic field gradient
shld - diamagnetic shielding
file - file reference
* - quit
The meaning of the four suboptions pot, fld, fldgrd and shld will probably present no
problems to you. For each of them, however, you will have to specify at which point(s) this
property should be calculated. This is accomplished by one or more data groups
$points in file control. After you chose one or more of the above options, you will
therefore reach the next submenu which deals with the specification of these data
groups:
there are 1 data groups $points
manipulate data group(s) $points
a - add another data group
m <integer> - modify <integer>th data group
m all - modify all data groups
d <integer> - delete <integer>th data group
d all - delete all data groups
off <integer> - switch off <integer>th data group
off all - switch off all data groups
on <integer> - switch on <integer>th data group
on all - switch on all data groups
s - scan through data groups
* - quit
The first line informs you how many of these data groups already exist in your control
file. Each of these data groups may consist of several points at which the properties will be
calculated. You may now create new data groups, delete old ones or simply switch on or
off individual data groups (without deleting them from control). The number of different
data groups $points as well as the number of points in each of them are not limited.
However, if you use many points, you should consider specifying them in a separate file.
This is most easily done using option file in the potential menu. This option will create
a file for your data groups $points and will write a reference of this file to file
control.
Option cowan-griffin
This option activates the computation of the first order relativistic correction to the
energy as given by the expectation value of the Cowan–Griffin operator.
Option localization
Specifying option localization will switch on a Boys localization of molecular
orbitals. define by default chooses a set of MOs to be localized according to a
certain threshold for the orbital energy. Information about these are displayed like
this:
BOYS localization will be performed with respect to x y z
number of sweeps = 10000
subset of molecular orbitals to be localized :
---> all occupied molecular orbitals
with orbital energy above -2.00000 Hartree
----------------------------------------------------------------
shells to be localized
----------------------------------------------------------------
a1 4-5 # 1- 5
e 2 # 1- 2
----------------------------------------------------------------
you are employing default options for localization
do you want to modify them ? DEFAULT(n)
If you want to change the MO selection or other options for the localization enter y at this
point (By default or when typing n you will reach the moloch options menu again). You
will then be asked whether to change the MO selection method. If you want this, you
will enter a little submenu where you can choose one of three possible selection
procedures:
-
all
-
selects all occupied orbitals
-
thr
-
selects all occupied orbitals with orbital energy larger than a certain
threshold
-
man
-
enables you to select the MOs manually later in this section
If the selection method thr is specified you then will be asked for the threshold to be
applied for the selection. Afterwards you have the possibility to change some other topics
concerning the localization:
- specify other localization directions
- switch on utilization of localized orbitals for population analysis and/or
preparation of plot data within the same moloch run
- set the maximum number of sweeps in the localization procedure
- specify a file where localized orbitals shall be written to
Option population analyses
When activating this option you first have to specify whether the population analysis (PA)
should be performed in the CAO (default) or AO basis. Afterwards define will ask you
whether you want to perform a Mulliken population analysis. In this case, the following
submenu will be displayed:
add or delete one or more special options for a
mulliken population analysis
option | status | description
-------|--------|---------------------------------------
spdf | F | compute MO contributions to atomic
| | brutto populations
molap | F | compute MO contributions to atomic
| | overlap populations
netto | F | compute atomic netto populations
irpspd | F | compute IRREP contributions to atomic
| | brutto populations
irpmol | F | compute IRREP contributions to atomic
| | overlap populations
mommul | F | print electrostatic moments resulting
| | from atomic charges
-------|--------|---------------------------------------
-<option> : switch off <option>
* or q(uit) : leave this menu
Here you can activate several optional quantities to be computed along with the Mulliken
PA. To switch on one or more of these options you must enter the corresponding option
keywords, e.g. spdf netto for computation of atomic neto populations and MO
contributions to atomic brutto populations. The status flags for these tasks will then
change from F (false) to T (true). To switch off any option you simply have to enter the
corresponding keyword preceded by a ‘-’, e.g. -netto for disabling calculation of atomic
netto populations.
After having left the Mulliken PA section you will be asked whether a population analysis
based on occupation numbers (a modified Roby–Davidson PA) should be performed
by moloch. When typing y you will see the following submenu, where you can
switch on several special options for the PA in the same manner as described
above.
add or delete one or more special options for a
population analysis based on occupation numbers
option | status | description
--------|--------|----------------------------------------
momao | F | compute MO contributions to modified
| | atomic orbital (MAO) occupation numbers
maodump | F | dump all MAOs onto standard output
maofile | F | write MAOs onto a separate file
select | F | write only those MAOs which have been
| | employed in the population analysis
all | F | write all MAOs
--------|--------|----------------------------------------
note that the options select and all are complementary
-<option> : switch off <option>
* or q(uit) : leave this menu
Afterwards you have the possibility to change the criterion to be applied for the selection
of modified atomic orbitals (MAOs) within the following little submenu:
global criterion for selection of Modified Atomic Orbitals (MAOs) :
-------------------------------------------------------------------
MAOs are employed if ’atomic’ density eigenvalues
exceed a threshold of .1000
-------------------------------------------------------------------
specify the appropriate option if you want to use another
global criterion for selecting MAOs
option | status | description
--------|--------|---------------------------------------
eig <r> | T | select by eigenvalues of the
| | ’atomic’ density matrices
occ <r> | F | select by occupation numbers
--------|--------|---------------------------------------
<r> is the selection threshold (DEFAULT= .1000 )
* or q(uit) : leave this menu
The criterion applied by default is the so-called atomic density eigenvalue with a threshold
of 0.1. You can switch the criterion to occupation numbers by entering occ. If you also
want to change the threshold, you just have to append its new value to the selection
keyword, e.g. occ .2. Finally you can select or disable various options in connection
with the computation of shared electron numbers (SEN) within the following
menu:
actual settings for data group $shared electron numbers
2-center shared electron numbers will be computed;
values are printed if absolute value exceeds .0100
3-center shared electron numbers will be computed;
values are printed if absolute value exceeds .0100
4-center shared electron numbers will be computed;
values are printed if absolute value exceeds .0100
add or delete one or more options for the
computation of Shared Electron Numbers (SEN)
option | status | description
--------|--------|----------------------------------------
2c <r> | T | compute 2-center SEN and print if
| | |SEN| > <r> (DEFAULT = .1000E-01)
3c <r> | T | compute 3-center SEN and print if
| | |SEN| > <r> (DEFAULT = .1000E-01)
4c <r> | T | compute 4-center SEN and print if
| | |SEN| > <r> (DEFAULT = .1000E-01)
--------|--------|----------------------------------------
nosym | F | switch off use of symmetry
orbs | F | compute orbital contributions to SEN
irreps | F | compute irrep contributions to SEN
--------|--------|----------------------------------------
-<option> : switch off <option>
* or q(uit) : leave this menu
The procedure for changing the options is the same as described above. By default
calculation of 2-, 3- and 4-center SENs will be enabled with thresholds of 0.01
each.
Option plot
This option allows you to prepare the data needed for contour plots of orbital amplitudes
or total electron densities. We do not recommend to prepare plotting data this way; an
easier method—with an easier syntax—is to generate these data directly by the programs,
where densities (also MP2 or excited ones) and Molecular orbitals are calculated. This is
described in Chapter 18. If you nevertheless want to prepare the input for plotting data
as needed by moloch using define, on activating plot you get the following
menu:
there are 1 data groups $grid
manipulate data group(s) $grid
a - add another data group
m <integer> - modify <integer>th data group
m all - modify all data groups
d <integer> - delete <integer>th data group
d all - delete all data groups
off <integer> - switch off <integer>th data group
off all - switch off all data groups
on <integer> - switch on <integer>th data group
on all - switch on all data groups
s - scan through data groups
* - quit
The commands in this menu serve for the manipulation of data groups $grid in an
analogous way as described for $points in the potential section above. $grid data groups
contain the input information necessary to create the plot data by moloch (one data
group for each plot). If you want to add a new data group you will enter this
submenu:
specify the input orbital / input density :
mo <label> - use occupied molecular orbital <label>
mo density - use one electron density built from the
occupied molecular orbitals
lmo <i> - use localized molecular orbital no. <lmo>
mao <i> <k> - use modified atomic orbital no. <i>
centered on atom no. <k>
help - explanation of the syntax for <label>
* - quit
Here you may specify the orbital to be plotted. To plot the amplitude of the fifth orbital in
irrep a1, e.g., you would enter mo 5a1. Equivalently you can use localized orbitals
from a Boys localization procedure or modified atomic orbitals as obtained in a
Roby–Davidson–Ahlrichs–Heinzmann population analysis. In the latter cases you will not
have to enter an irrep label, as these orbitals are necessarily in C1 symmetry. Instead you
will have to enter the index of the orbital to be plotted (and for option mao the index of
the atom at which it is situated). In all cases you will additionally have to specify
the plane in which the amplitudes or densities will be monitored. To do this,
you have to declare two vectors which span that plane and the origin of this
new coordinate system relative to the one in which the atomic coordinates are
given. Furthermore, you will have to create a grid of points on this plane. The
orbital amplitude or electron density will then be calculated for every point in
this grid. The grid is created by telling define the range to be included along
both vectors spanning the plane (where the unit in each direction is the length
of the corresponding basis vector) and the number of points to be calculated
in this range. It is advantageous to use a wide grid while you test the ranges
or planes which give the best results and then to switch to a finer grid for the
final calculation. Finally input (MO vector) and output (plot data) files can be
specified.
In case you do not want to add a new data group as described above but to change
an existing one, you will be asked which one of the specifications you want to
modify.
![[ ]
χμ = (x - x0)l(y - y0)m (z - z0)n exp - ημ(r - r0)2](DOK2x.png)
![χ = (x - x )l(y - y )m(z - z )n exp[- αη (r - r )2]
μ 0 0 0 μ 0](DOK3x.png)