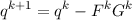
relax drives and controls a non-linear optimization procedure to locate the minimum (or a stationary point) of a function f(x). In TURBOMOLE f is always the electronic energy, and the coordinates x will be referred to as general coordinates. They include
The optimization employs an iterative procedure based on gradients ∇f of the current and, if available, previous iterations. Various procedures can be applied: steepest descent, Pulay’s DIIS, quasi–Newton, conjugate gradients, as well as combinations of them. relax carries out:
The mode of operation is chosen by the keywords $optimize and $interconversion and the corresponding options, which will be described in the following sections.
After gradients Gk have been calculated for coordinates qk in optimization cycle k, new coordinates (or basis set exponents) qk+1 can be obtained from the quasi–Newton update:
|
|
where Fk is the inverse of an approximate force constant matrix Hk. This method would immediately converge to the equilibrium geometry if Fk would be the inverse of the exact force constant matrix and the force field would be quadratic. In real applications usually none of these requirements is fulfilled. Often only a crude approximation to the force constant matrix Hk is known. Sometimes a unit matrix is employed (which means coordinate update along the negative gradient with all coordinates treated on an equal footing).
The optimization of nuclear coordinates in the space of internal coordinates is the default task performed by relax and does not need to be enabled. Any other optimization task requires explicit specifications in data group $optimize, which takes several possible options:
Structure optimization in internal coordinates.
Structure optimization in redundant coordinates.
Structure optimization in cartesian coordinates.
Optimization of basis set exponents, contraction coefficients, scaling factors.
Optimization of global scaling factor for all basis set exponents.
Note: All options except internal are switched off by default, unless they have been activated explicitly by specifying on.
Some of the options may be used simultaneously, e.g.
Other options have to be used exclusively, e.g.
The update of the coordinates may be controlled by special options provided in data group $coordinateupdate which takes as options:
Maximum total coordinate change (default: 0.3).
Calculate coordinate update by inter/extrapolation using coordinates and gradients of the last two optimization cycles (default: interpolate on) if possible.
Display optimization statistics for the integer previous optimization cycles. Without integer all available information will be displayed. off suppresses optimization statistics.
The following data blocks are used by program relax:
cartesian atomic coordinates and their gradients.
exponents and scale factors and their gradients.
global scale factor and its gradient.
cartesian atomic coordinates and their gradients.
global scale factor and its gradient.
the force constant matrix in the space of cartesian coordinates.
cartesian atomic coordinates.
exponents and scale factors.
global scale factor.
For structure optimizations the use of (redundant) internal coordinates is recommended, see Section 4.0.6. Normally internal coordinates are not used for input or output by the electronic structure programs (dscf, mpgrad, etc.). Instead the coordinates, gradients, etc. are automatically converted to internal coordinates by relax on input and the updated positions of the nuclei are written in cartesians coordinates to the data group $coord. Details are explained in the following sections.
In a Newton-type geometry update procedure often only a crude approximation to the force constant matrix Hk is available. What can be done then is to update Fk = (Hk)-1 in each iteration using information about previous coordinates and gradients. This constitutes the quasi–Newton or variable metric methods of which there are a few variants:
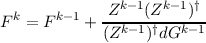 |
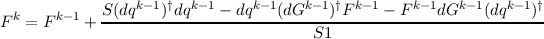 |
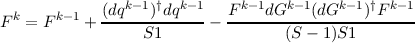 |
The meaning of the symbols above is as follows:
approximate inverse force constant matrix in the k-th iteration.s
general coordinates in the k-th iteration.
gradients in the k-th iteration.
An alternative is to use update algorithms for the hessian Hk itself:
Ehrig, Ahlrichs : Diagonal update for the hessian by means of a least squares fit
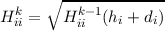 |
with the new estimate h for the diagonal elements obtained by
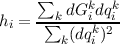 |
and the error d obtained by the regression
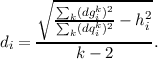 |
Another alternative is to use DIIS-like methods: structure optimization by direct inversion in the iterative subspace. (See ref. [37] for the description of the algorithm). The DIIS procedure can often be applied with good success, using static or updated force constant matrices.
Any of the algorithms mentioned above may be chosen. Recommended is the macro option ahlrichs, which leads to the following actions (n is the maximum number of structures to be included for the update, default is n = 4):
geometry update by inter/extrapolation using the last 2 geometries.
diagonal update for the hessian as described above; DIIS–like update for the geometry.
BFGS-type update of the hessian and quasi–Newton update of (generalized) coordinates.
References for the algorithms mentioned above: [34,37–41]
If structure optimizations are to be performed in the space of internal coordinates ($optimize internal, is the default setting), appropriate internal coordinate definitions have to be provided on data block $intdef. The types available and their definitions are described in Section 4.1.2. For recommendations about the choice of internal coordinates consult ref. [28]. Nevertheless the structure of $intdef will shortly be described. The syntax is (in free format):
The first items have been explained in Chapter 4.
Two additional items val=real, fdiag=real may be supplied for special purposes:
serves for the input of values for internal coordinates for the interconversion internal → cartesian coordinates; it will be read in by relax if the flag for interconversion of coordinates has been activated ($interconversion on ), or by the interactive input program define within the geometry specification menu.
serves for the input of (diagonal) force constants for the individual internal coordinates to initialize $forceapprox.
This is the default task of relax ($optimize internal on does not need to be specified!) You need as input the data groups :
cartesian coordinates and gradients as provided and accumulated in subsequent optimization cycles by the programs grad, or rdgrad etc.
definitions of internal coordinates.
definitions of redundant coordinates.
Output will be the updated coordinates on $coord and the updated force constant matrix on $forceapprox. If any non-default force constant update option has been chosen, relax increments its counting variables <numgeo>, <numpul> within command keyword $forceupdate. If the approximate force constant has been initialized ($forceinit on ) relax switches the initialization flag to $forceinit off. Refer also to the general documentation of TURBOMOLE. It is recommended to check correctness of your definition of internal coordinates:
For this task you have to specify:
These lines switch on the non-default optimization in cartesian coordinates and switch off the optimization in internal coordinates (this has to be done explicitly!). As input data groups you need only $grad as provided by on of the gradient programs. For the first coordinate update an approximate force constant matrix is needed in data group $forceapprox. Output will be the updated coordinates on $coord, and the updated force constant matrix on $forceapprox.
The coordinates for any single atom can be fixed by placing an ’f’ in the third to eighth column of the chemical symbol/flag group. As an example, the following coordinates specify acetone with a fixed carbonyl group:
For this task you have to specify:
This example would perform only a basis set optimization without accompanying geometry optimization. It is possible, of course, to optimize both simultaneously: Just leave out the last line of the example (internal off). Input data groups are:
Basis set exponents, contraction coefficients, scaling factors and their respective gradients as provided and accumulated in subsequent optimization cycles by one of the programs grad or mpgrad, if $drvopt basis on has been set.
Description of basis sets used, see Section 4.2.
Output will be the updated basis on $basis, and the updated force constant matrix on $forceapprox.
For an example, see Section 21.5.
The optimization of geometry and basis set may be performed simultaneously and requires the specification of:
and needs as input data groups $grad and $egrad. Output will be on $coord, $basis, also on $forceapprox (updated).
Optimization of a global scaling factor is usually not performed in geometry optimizations. It is a special feature for special applications by even more special users. As reference see [42].
To optimize the structure and a global scaling factor specify:
You need as input data groups $grad and $globgrad, the latter contains the global scaling factors and their gradients accumulated in all optimization cycles. Output will be on $coord, $global, also on $forceapprox (updated). Note that for optimization of a global scaling factor a larger initial force constant element is recommended (about 10.0).
Due to translational and rotational degrees of freedom and the non-linear dependence of internal coordinates upon cartesian coordinates, there is no unique set of cartesian coordinates for a given set of internal coordinates. Therefore an iterative procedure is employed to calculate the next local solution for a given cartesian start coordinates. This task may be performed using the relax program, but it is much easier done within a define session.
To perform this tasks, you have to activate the interconversion mode by
Note that any combination of the three options showed is allowed! The default value is coordinate, the two other have to be switched on explicitly if desired.
You need as input data groups:
Definitions of (redundant) internal coordinates
Cartesian coordinates (for option ‘coordinate’)
Cartesian coordinates and gradients as provided and accumulated in subsequent optimization cycles by the various gradient programs (for coordinate and gradient)
Analytical force constant matrix (as provided by the force constant program aoforce) (only if option hessian is specified). The data group $hessian (projected) may be used alternatively for this purpose.
All output will be written to the screen except for option hessian (output to data group $forceapprox)
The m-matrix serves to fix position and orientation of your molecule during geometry optimizations. It cannot be used to fix internal coordinates! The m-matrix is a diagonal matrix of dimension 3n2 (where n is the number of atoms). Normally m will be initialized as a unit matrix by relax. As an example consider you want to restrict an atom to the xy-plane. You then set the m(z)–matrix element for this atom to zero. You can use at most six zero m-matrix diagonals (for linear molecules only five)—corresponding to translational and rotational degrees of freedom. Note that the condition of the BmB†-matrix can get worse if positional restrictions are applied to the m-matrix. m-matrix elements violating the molecular point group symmetry will be reset to one. Non-default settings for m-matrix diagonals of selected atoms have to be specified within data group $m-matrix as:
The most simple initial hessian is a unit matrix. However, better choices are preferable. For structure optimizations using internal coordinates you may use structural information to set up a diagonal force constant matrix with elements chosen in accord to the softness or stiffness of the individual modes. For detailed information refer to ref. [40]. For optimization of basis set parameters less information is available. When neither data block $forceapprox is available nor $forceinit on is set, the force constant matrix will be initialized as a unit matrix. Specifying the force constant initialization key $forceinit on diag=... will lead to:
Initialization with real as diagonal elements.
Initial force constant diagonals will be assigned the following default
values:
| internal coordinates | : | stretches | 0.50 |
| angles | 0.20 | ||
| scaling factors | : | s,p | 1.50 |
| d | 3.00 | ||
| exponents | : | uncontracted | 0.15 |
| contracted | 10.00 | ||
| contraction coefficients | : | 100.00 | |
| global scaling factor | : | 15.00 | |
| cartesian force constants | : | 0.50 | |
Initial force constant diagonals will be taken from
$intdef fdiag=... or
$global fdiag=...
Similar initialization modes are NOT supported for geometry
optimization in cartesian space and for the optimization of basis set
parameters!
The energy file includes the total energy of all cycles of a structure optimization completed so far. To get a display of energies and gradients use the UNIX command grep cycle gradient which yields, e.g. H2O.
This should be self-evident. To see the current—or, if the optimization is converged, the final—atomic distances use the tool dist. Bond angles, torsional angles etc. are obtained with the tools bend, tors, outp, etc. In the file gradient are the collected cartesian coordinates and corresponding gradients of all cycles. The values of the general coordinates and corresponding gradients are an output of relax written to job.<cycle> of job.last within jobex. To look at this search for ‘Optimization statistics’ in job.last or job.<cycle>.