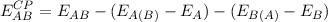
The shell script jobbsse controls and executes the automatic calculation of the counterpoise correction as it has been formulated by Boys and Bernadi (S. F. Boys and F. Bernardi, Mol. Phys., 19, 553 (1970)) to estimate the Basis Set Superposition Error (BSSE). For a dimer, the cp-correction takes the form for the monomers A and B:
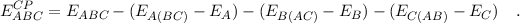
Note that you need to set up the fragments (and possibly their symmetries using define in the geometry menu beforehand. The general structure of a jobbsse calculation is as follows:
Note, that counterpoise-corrected calculations with jobbsse are NOT as black-box as ordinary geometry optimizations with jobex. The input generated for the fragments are based on the default occupation numbers obtained from the EHT guess, default assignments for the frozen orbitals, memory, etc. Since this might be different from what is needed (or even fail), it is recommended to let jobbsse stop after the initial setup step using the flag -setup and to check carefully the assigned basis sets, occupations number and subsystem symmetries. In particular, for MP2 or CC2 calculations with molecules containing not only the atoms H–Ar also the number of frozen orbitals should be checked, and if neccessary corrected.
Given a shell the usage is:
nohup jobbsse &
This command invokes cp-correction, and, if needed structure optimization using the default program statpt. Note, that the program needs to know which calculation is being done. Structure optimizations using program relax can be performed using -relax flag:
nohup jobbsse -opt -relax &
nohup means that the command is immune to hangups, logouts, and quits. & runs a background command. jobbsse accepts the following arguments controlling the level of calculation, convergence criteria and many more (for example nohup jobbsse -gcart 4 &):
converge total energy up to
10(-<integer>) Hartree (default: 6)
converge maximum norm of cartesian gradient up to
10(-<integer>) atomic units (default: 3)
perform up to integer cycles (default: 100)
calculate the gradient as well
optimise the structure
define the optimization level, level=scf, dft, mp2, or cc2 (default is scf). Note that the program needs this input! If the level is DFT, the grid will be automatically set to m4.
use RI modules ridft and rdgrad (fast Coulomb approximation) instead of dscf and grad as well as rimp2 instead of mpgrad
employ programs from directory <path>
Is able to control the memory from outside define Note that if you did not define any memory, it is automatically set to 1 GB
calculates, in case we have a trimer:
Energy = ABC - AB(C) + AB - AC(B) + AC - BC(A) +
BC
rather than
Energy = ABC - A(BC) + A - B(AC) + B - C(AB) + C
(note that the first term neglects the BSSE in the dimer)
Interrupt calculation after the initial setup step to check and possibly correct the control files for the fragments and the supermolecule. To continue, start jobbsse without the -setup option.
shows a short description of the commands above
There will be an output written to file bsse_out. In this file, you will find all individual energies computed which were used to calculate the last cp-corrected energy. The same holds true for the last gradients, which are written to grad_out.
The convergence criteria and their current values are written out at the not.converged file. For the possible options to control convergence check the subsection for the optimization program used (statpt, which is used by default, or relax). Since for weak complexes the force constants for intra- and intermolecular bonds very strongly in magnitude, it is recommended to use whenever possible redundant internal coordinates.