9.3 How to Prepare and Perform MP2 Calculations
Prerequisites
Calculations with mpgrad, ricc2 or pnoccsd require
- a converged SCF calculation with the one-electron density convergence threshold set
to $denconv 1.d-7 or less
- the maximum core memory the program is allowed to allocate should be defined in
the data group $maxcor (in MB); the recommended value is ca. 3/4 of the available
(physical) core memory at most.
- orbitals to be excluded from the correlation treatment have to be specified in the data
group $freeze
- for mpgrad the calculation of gradients is omitted by adding the flag $mp2energy to
the control file; in this case only the MP2 energy is calculated.
Calculations with ricc2 and pnoccsd moreover use
- an auxiliary basis defined in the data groups $atoms and $cbas. If not specified before
the start of ricc2 or pnoccsd the programs will assign the default auxiliary basis
sets which have the same names as the one-electron basis sets and will try to extract
them from the TURBOMOLE basis set library. If this succeeds the assignment and
auxiliary basis sets are stored in the control file else the programs will stop.
This is not needed for mpgrad, but here one needs
- a specification for scratch files and their size in data group $mointunit (see
Section 20.2.18)
- and the number of passes for integral evaluations and transformations in data group
$traloop
For explicitly-correlated MP2-F12 calculations one needs—depending the details of the applied
approximations—additionally a so-called complementary auxiliary basis set (CABS, defined in
$cabs) and a RI-SCF auxiliary basis set defined in $jkbas.
Calculations with ricc2 and pnoccsd
-
1.
- RI-MP2 calculations require the specification of auxiliary basis sets ($cbas) and a
converged SCF calculation with the one-electron density convergence threshold set to
$denconv 1.d-7 or less. In addition, the options $freeze (frozen core approximation)
and $maxcor (maximum core memory usage) should be set. All these settings can be
done during the input generation with the program define under the entries mp2/cc
or pnocc of the last main menu.
-
2.
- The ricc2 program requires the data group:
$ricc2
mp2
geoopt model=mp2
(Where the last line should only be included if the calculation of gradients is needed
e.g. in geometry optimizations.) This can be prepared with define in the menu cc.
-
3.
- The pnoccsd program does not require any special input for a MP2 calculation with
default thresholds but it is recommended to specify the PNO truncation threshold
explicitly in the data group:
$pnoccsd
mp2
tolpno= 1.0E-7
This can be prepared with define in the menu pnocc.
-
4.
- For explicitly-correlated MP2-F12 calculations with ricc2 or pnoccsd also the data
groups $rir12 and $lcg are needed.
-
5.
- Start a single ricc2 and pnoccsd calculations respectively with the commands ricc2
and pnoccsd.
-
6.
- For optimisation of structure parameters at the RI-MP2 level with ricc2 use
the command jobex -level cc2. For geometry optimizations with RI-JK-SCF as
reference for RI-MP2 with the ridft and ricc2 binaries the additional option -rijk
has to be given.
-
7.
- The combination of RI-MP2 with RI-JK-SCF can lead to significant computational
savings in particular for geometry optimizations for small and medium sized molecules
with large basis sets (quadruple-ζ and beyond) or basis sets with diffuse functions
(e.g. the aug-cc-pVXZ basis set families). For large molecules with TZVPP or similar
basis sets, conventional direct SCF calculations are usually more efficient.
-
8.
- With ricc2 and pnoccsd spin-component scaled SCS- or SOS-RI-MP2 calculations can be
carried out by adding in the $ricc2 (for ricc2) or $pnoccsd (for pnoccsd data group the
line
scs cos=1.2d0 css=0.3333d0
where the two parameters are the scaling factors for, respectively, the opposite- and
same-spin contribution. The specification of the scaling factors is optional; the
default values are cos=6/5 and css=1/3 as recommended by S. Grimme in J.
Chem. Phys. 118 (2003) 9095. The abbreviation sos can be used for SOS-MP2
calculations with cos=1.3 and css=0.0 (Y., Jung, R.C. Lochan, A.D. Dutoi, and
M. Head-Gordon, J. Chem. Phys. 121 (2004) 9793.). SOS-MP2 is in ricc2 implemented
with 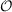 (
(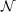 4) scaling costs. For such calculations the data group $laplace has to be
added.
4) scaling costs. For such calculations the data group $laplace has to be
added.
-
9.
- For technical recommendations and additional options for parallel RI-MP2 and PNO-MP2
calculations with the ricc2 and pnoccsd programs see Secs. 3.2 and 10.6 and
9.7.
MP2 calculations with a ROHF reference state
With the program ricc2 it is possible to compute MP2 (and spin-component scaled) MP2 energies
with single-determinant restricted open-shell reference wavefunctions. No additional input is
required apart from the usual ROHF input for the dscf and ridft programs and a standard MP2
input for ricc2.
TURBOMOLEs Hartree-Fock codes can handle within the ROHF framework many cases,
which include beside common high- and low-spin configuration state functions also
weighted averages of high-spin CSFs (see Sec. 6.3 for further details). The Møller-Plesset
perturbation theory and coupled cluster functionalities implemented in ricc2 require a
single-determinant reference state and can thus only deal with high-spin open-shell cases (not
averaged):
- The spins of all nopen unpaired electrons are parallel (α spin will be assumed) so that
the ROHF reference state has the spin multiplicity nopen + 1.
- There must be only one type of open shells and all orbitals in this shell must have the
occupation number 1.
- For the single electron case (i.e. doublets) the Roothaan parameters are a = b = 0,
for high-spin cases with more than one unpaired electron the Roothaan parameters
must be set to a = 1 and b = 2.
For non-abelian points groups this implies that shells with degenerate orbitals (as e.g. t1 in
point group I) must be half-filled. An average over the different (symmetry-equivalent
or inequivalent) high-spin determinants that are obtained when a shell of degenerate
orbitals is less than or more than half-filled is not possible with single-point ricc2
calculations.
For states with less than or more than half-filled shells of degenerate orbitals the calculations half
to be done in a point group that lifts the degeneracy such that it becomes possible to assign
integer occupation numbers. A symmetry-breaking of the orbitals can be avoided by doing the
Hartree-Fock calculation in the full point group. The input (and MO coefficients) can then be
transformed to a lower point group using define only for the ricc2 calculation.
Calculations with mpgrad
-
1.
- Add $denconv 1.d-7 to the control file and perform a dscf run.
-
2.
- If any orbitals are decided to be excluded from MP2 treatment, add data group
$freeze manually to the control file, see also Section 20.2.18.
-
3.
- For preparation of an mpgrad run use the script Mp2prep:
mp2prep -e/g -m memory -p discspace [scratch file directory]
As an example, with the command
mp2prep -e -m 100 -p 1000 /work
an MP2-energy calculation is prepared, the amount of available core memory is
restricted to 100 MB, the MOs are blocked, so that integral scratch files—located
in the directory /work—do not need more than 1000 Mb. The number of blocks,
i.e. the number of passes with repeated integral evaluations, is written to the
control file ($traloop) as well as the specification of scratch files ($mointunit, see
Section 20.2.18). Note: less disc space means more passes and thus lower efficiency
of mpgrad, but due the technical limitations discspace should be limited to values <
16Gb to avoid integer overflow errors. Settings obtained by mp2prep may be changed
manually. You may change the number of passes in $traloop by editing the control
file (e.g. if the originally intended disc space is not available). To adapt the size of
scratch files add $statistics mpgrad to control file and start an mpgrad statistics
run with the command mpgrad.
-
4.
- Start a single mpgrad calculation with the command mpgrad.
-
5.
- For optimisation of structure parameters at the (non-RI-) MP2 level use the command
jobex -level mp2. Note, that the frozen core approximation is ignored in this case.