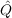 12f12|ij⟩,
12f12|ij⟩,To obtain the F12 correction to the MP2 energy, the data group $rir12 must be added to the control file. A typical run will include the input:
The MP2-F12 ground-state energy is
| EMP2-F12 | = EMP2 + EF12, | (9.5) |
| 12f12|ij⟩, | (9.6) |
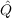 12 defines the doubles
excitation space covered by the geminals (it also ensures strong orthogonality to the
occupied orbitals). Usually
12 defines the doubles
excitation space covered by the geminals (it also ensures strong orthogonality to the
occupied orbitals). Usually 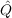 12 is chosen to be
12 is chosen to be 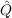 12 = (1 -Ô1)(1 -Ô2) -
12 = (1 -Ô1)(1 -Ô2) -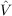 1
1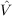 2, where
Ôμ = ∑
k|φk(μ)⟩⟨φk(μ)| is the projection operator onto the space spanned by the occupied
spin orbitals φk and
2, where
Ôμ = ∑
k|φk(μ)⟩⟨φk(μ)| is the projection operator onto the space spanned by the occupied
spin orbitals φk and 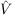 μ = ∑
a|φa(μ)⟩⟨φa(μ)| is the projector onto the virtual spin
orbitals.
μ = ∑
a|φa(μ)⟩⟨φa(μ)| is the projector onto the virtual spin
orbitals.
The F12 correction is obtained by minimizing the functional
| FF12 | = ∑
i<j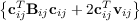 | (9.7) |
| vij(kl) | = ⟨kl|f12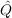 12r12-1|ij⟩, 12r12-1|ij⟩, | (9.8) |
| Bij(kl,mn) | = ⟨kl|f12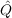 12( 12(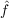 1 + 1 + 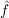 2 - εi - εj) 2 - εi - εj)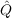 12f12|mn⟩, 12f12|mn⟩, | (9.9) |
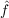 μ is the Fock operator for electron μ and εk is a (semi-)canonical Hartree–Fock orbital
energy.
μ is the Fock operator for electron μ and εk is a (semi-)canonical Hartree–Fock orbital
energy.
A MP2-F12 calculation is defined through a number of choices concerning the nature of the
geminals (f12 and 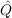 12), the geminal excitation space (ijkl or ijij) and approximations in
computing the B matrix (GBC, EBC, [
12), the geminal excitation space (ijkl or ijij) and approximations in
computing the B matrix (GBC, EBC, [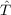 ,f12]). These choices correspond to keywords in the
$rir12 data group, explained below.
,f12]). These choices correspond to keywords in the
$rir12 data group, explained below.
To run a MP2-F12 calculation, one has to select the auxiliary basis sets cbas, cabs and optionally jkbas. The ricc2 program uses the robust fitting techniques of Ref. [135] for the F12 integrals and the cbas basis is used for both the F12 and the usual MP2 Coulomb integrals. For the density fitting of the Coulomb and exchange matrices of the Fock matrix, the jkbas will be used instead of the cbas basis if it is included in the control file (this is recommended and is achieved using the rijk menu in define). For the RI approximation of the 3- and 4-electron integrals as sums of products of 2-electron integrals, intrinsic to the F12 method, the complementary auxiliary basis (CABS) approach is used [136]. If define is used to set up the cabs basis, the library cabasen is searched. This library contains the optimised cabs basis sets [137] for the cc-pVXZ-F12 basis sets of Peterson et al. [138]. For other basis sets, the auxilliary basis in the library cabasen is identical with the auxilliary basis in the library cbas.
The $rir12 data group may be set by choosing the f12 option in the cc menu when running define. This command activates the f12 menu, where the default options may be changed if desired:
corresponds to the choice of 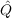 12. Almost all modern MP2-F12
calculations use ansatz 2 (default), which gives much improved energies
over ansatz 1 (see Ref. [139] for details). The principal additional cost of
using ansatz 2 over ansatz 1 is concerned with the coupling between the
F12 and conventional amplitudes. This is avoided by choosing 2*, which
corresponds to neglecting EBC (Extended Brillouin Condition) terms in
the Fock matrix elements.
12. Almost all modern MP2-F12
calculations use ansatz 2 (default), which gives much improved energies
over ansatz 1 (see Ref. [139] for details). The principal additional cost of
using ansatz 2 over ansatz 1 is concerned with the coupling between the
F12 and conventional amplitudes. This is avoided by choosing 2*, which
corresponds to neglecting EBC (Extended Brillouin Condition) terms in
the Fock matrix elements.
is the method of computing the matrices Bij (see Ref. [139] for details). The cost and accuracy increases from A to B. It is recommended to use B (default). The energies computed using A are then also printed out in the output.
is the method for approximately computing the integrals for the operator
[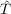 ,f12], where the matrix representations of F+K or T+V are used. F+K
(the core Hamiltonian plus Coulomb term) is recommended and is the
default.
,f12], where the matrix representations of F+K or T+V are used. F+K
(the core Hamiltonian plus Coulomb term) is recommended and is the
default.
refers to the method of orthogonalising the orbitals in the complementary auxiliary basis. Singular-value decomposition (svd) or Cholesky decomposition (cho) are available. svd is recommended and is the default, with a threshold of 1.0d-08. The basis set used for CABS is set from the cc menu.
refers to the choice of excitation space. inv is the orbital-invariant merhod of Ref. [140], with amplitudes cij(kl). noinv is the original orbital-dependent diagonal "ijij" method of Ref. [140], with amplitudes cij(ij) (not recommended, unless in combination with localised orbitals). fixed is the (diagonal and orbital-invariant) rational generator approach of Ref. [141], where the F12 amplitudes are not optimised but predetermined using the coalescence conditions (default). An additional keyword noflip suppresses the use of spin-flipped geminals in open-shell calculations; by default spin-flipped geminals are used as described in Ref. [19].
controls which orbitals are used in the F12 energy contribution. hf means that (semi-)canonical Hartree–Fock orbitals are used (default). rohf means that ROHF orbitals are used (any frozen orbitals will then also implicitly be ROHF). For calculations on closed-shell systems, localised orbitals may be used. Both the Boys [142] and Pipek–Mezey [143] methods are available for localisation of the orbitals.
corresponds to the choice of correlation factor f12 in the geminal basis functions. R12 results in a calculation using linear-r12 and LCG results in a calculation using the Slater-type correlation factor with exponent 1.4 a0-1, represented as a linear combination of six Gaussians (see Ref. [144]). Note that the exponents 0.9, 1.0 and 1.1 a0-1 are recommended for use with the cc-pVXZ-F12 basis sets [138].
switches on/off the calculation of a second-order correction to the Hartree–Fock energy by accounting for single excitations into the complementary auxiliary basis set (CABS). The single excitations into the CABS basis can be computed without extra costs if the CABS Fock matrix elements are required anyway for the F12 calculation (i.e., for ansatz 2, approximation B or comaprox F+K). The computation of CABS singles cannot be switched off if it is free of costs.
controls whether or not the F12 contribution to the MP2 pair energies appear in the output (default off).
Further options:
corrfac LCG refers to a further data group for the definition of the correlation factor. When define is used, the default is
The nature of the LCG correlation factor may be changed by editing this data group in the control file. For example, to use a Slater-type correlation factor with exponent 1.0 a0-1, represented as a linear combination of three Gaussians, use
Alternatively, the exponents and coefficients of the fit may be given explicitly:
MP2-F12 calculations may be combined with Grimme’s SCS approach (S. Grimme, J. Chem. Phys. 118 (2003) 9095) by inserting scs in $ricc2,
In this case, the SCS parameters cos=6/5 and css=1/3 are used. Also individual scaling factors for the same-spin and opposite-spin contributions may be defined, see Section 10.7.
For open-shell calculations, two choices of the examp fixed method are available. These are controled by a keyword in the $rir12 data group
These differ in the treatment of the αβ block, where either only the diagonal excitations enter (with amplitude 0.5) diag, or the equivalent of the spin-adapted singlet and triplet pair excitations enter (as far as possible) full. Note that the diag method with UMP2-F12 yields a result different to that of fixed MP2-F12, even for identical RHF and UHF determinants. However, the diag method is somewhat less expensive than the full method.
Recommendations for orbital and auxiliary basis sets:
The best orbital basis sets to use for MP2-F12 calculations are probably the cc-pVXZ-F12 basis sets, specially optimised for MP2-F12 calculations [138] for the atoms H, He, B–Ne and Al–Ar. In conjunction with these cc-pVXZ-F12 basis sets, we recommend to use the optimised cc-pVXZ-F12 sets of Yousaf and Peterson [137] as cabs. Furthermore, cbas and jkbas basis sets can be selected from the cbasen and jkbasen libraries, respectively, using the alias cc-pVXZ-F12 (a jkbas is currently not available for He, Ne and Ar). This alias points to the corresponding aug-cc-pwCV(X+1)Z cbas and aug-cc-pV(X+1)Z jkbas. These recommendations are on the side of caution and are likely to be refined as more experience is gained [145–147].
For atoms other than H, He, B–Ne and Al–Ar, optimised F12 basis sets are not yet available. In this case, basis sets must be selected and/or optimised carefully. It is advised to contact the Theoretical Chemistry Group in Karlsruhe for support (e-mail to: klopper@kit.edue).