 ∑
aibjtabijτ
aibj .
∑
aibjtabijτ
aibj .In CCSD the ground–state energy is (as for CC2) evaluated as
| ECC | = ⟨HF|H|CC⟩ = ⟨HF|H exp(T)|HF⟩ , | (11.1) |
| T1 | = ∑ aitaiτai , | (11.2) |
| T2 | = ∑
aibjtabijτ
aibj . | (11.3) |
| Ωμ1 | = ⟨μ1|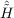 + [ + [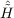 ,T2]|HF⟩ = 0 , ,T2]|HF⟩ = 0 , | (11.4) |
| Ωμ2 | = ⟨μ2|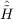 + [ + [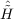 ,T2] + [[Ĥ,T2],T2]|HF⟩ = 0 , ,T2] + [[Ĥ,T2],T2]|HF⟩ = 0 , | (11.5) |
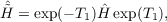
| EMP3,tot = | EHF + EMP2 + EMP3 | (11.6) |
| = | ⟨HF|Ĥ + [Ĥ,T2(1)]|HF⟩ + ∑ μ2tμ2(1)⟨μ 2|[Ŵ,T2(1)]|HF⟩ | (11.7) |
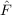 . To evaluate the fourth-order energy one needs in addition to the first-order also
the second-order amplitudes, which are obtained from the solution of the equations
. To evaluate the fourth-order energy one needs in addition to the first-order also
the second-order amplitudes, which are obtained from the solution of the equations
⟨μ1|[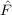 ,T1(2)] + [Ŵ,T
2(1)]|HF⟩ = ,T1(2)] + [Ŵ,T
2(1)]|HF⟩ = | 0 | (11.8) |
⟨μ2|[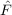 ,T2(2)] + [Ŵ,T
2(1)]|HF⟩ = ,T2(2)] + [Ŵ,T
2(1)]|HF⟩ = | 0 | (11.9) |
⟨μ3|[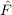 ,T3(2)] + [Ŵ,T
2(1)]|HF⟩ = ,T3(2)] + [Ŵ,T
2(1)]|HF⟩ = | 0 | (11.10) |
| EMP4 = | ∑ μ2tμ2(1)⟨μ 2|[Ŵ,T1(2) + T 2(2) + T 3(2)] + [[Ŵ,T 2(1)],T 2(1)]|HF⟩ . | (11.11) |
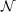 is a measure for the system
size (e.g. the number of atoms), the computational costs (in terms of floating point
operations) for CCSD calculations scale as
is a measure for the system
size (e.g. the number of atoms), the computational costs (in terms of floating point
operations) for CCSD calculations scale as 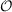 (6). If for the same molecule the number of
one-electron basis functions N is increased the costs scale with (4). (For RI-MP2 and
RI-CC2 the costs scale with the system size as (5) and with the number of basis
functions as (3).) The computational costs for an MP3 calculations are about the
same as for one CCSD iteration. For MP4 the computational costs are comparable to
those for two CCSD iteration plus the costs for the perturbation triples correction (see
below).
(6). If for the same molecule the number of
one-electron basis functions N is increased the costs scale with (4). (For RI-MP2 and
RI-CC2 the costs scale with the system size as (5) and with the number of basis
functions as (3).) The computational costs for an MP3 calculations are about the
same as for one CCSD iteration. For MP4 the computational costs are comparable to
those for two CCSD iteration plus the costs for the perturbation triples correction (see
below).
Explicitly-correlated CCSD(F12) methods:
In explicitly-correlated CCSD calculations the double excitations into products of virtual
orbitals, described by the operator T2 = 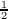 ∑
aibjtabijτaibj, are augmented with double
excitations into the explicitly-correlated pairfunctions (geminals) which are described in
Sec. 9.5:
∑
aibjtabijτaibj, are augmented with double
excitations into the explicitly-correlated pairfunctions (geminals) which are described in
Sec. 9.5:
| T | = T1 + T2 + T2′ | (11.12) |
| T2′ | =  ∑
ijklcijklτ
kilj ∑
ijklcijklτ
kilj | (11.13) |
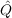 12f12|kl⟩ (for the defintion
12f12|kl⟩ (for the defintion 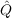 12 and f12 see Sec. 9.5). This enhances
dramatically the basis set convergence of CCSD calculations ( [17]). Without any further
approximations than those needed for evaluating the neccessary matrix elements, this extension of
the cluster operator T leads to the CCSD-F12 method. CCSD(F12) is an approximation
( [17,162]) to CCSD-F12 which neglects certain computationally demanding higher-order
contributions of
12 and f12 see Sec. 9.5). This enhances
dramatically the basis set convergence of CCSD calculations ( [17]). Without any further
approximations than those needed for evaluating the neccessary matrix elements, this extension of
the cluster operator T leads to the CCSD-F12 method. CCSD(F12) is an approximation
( [17,162]) to CCSD-F12 which neglects certain computationally demanding higher-order
contributions of 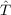 2′. This reduces the computational costs dramatically, while the accuracy of
CCSD(F12) is essentially identical to that of CCSD-F12 [163,164]. In the CCSD(F12)
approximation the amplitudes are determined from the equations:
2′. This reduces the computational costs dramatically, while the accuracy of
CCSD(F12) is essentially identical to that of CCSD-F12 [163,164]. In the CCSD(F12)
approximation the amplitudes are determined from the equations:
| Ωμ1 | = ⟨μ1|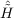 + [ + [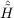 ,T2 + T2′]|HF⟩ = 0 , ,T2 + T2′]|HF⟩ = 0 , | (11.14) |
| Ωμ2 | = ⟨μ2|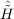 + [ + [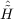 ,T2 + T2′] + [[Ĥ,T2 + 2T2′],T2]|HF⟩ = 0 , ,T2 + T2′] + [[Ĥ,T2 + 2T2′],T2]|HF⟩ = 0 , | (11.15) |
| Ωμ2′ | = ⟨μ2′|[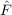 ,T2′] + ,T2′] + 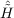 + [ + [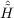 ,T2]|HF⟩ = 0 . ,T2]|HF⟩ = 0 . | (11.16) |
| ECCSD(F12)-SP | = LCCSD(F12) = ⟨HF|H|CC⟩ + ∑ μ2′cμ2′Ωμ2′ | (11.17) |
The SP approach becomes in particular very efficient if combined with the neglect of certain higher-order explicitly-correlated contributions which have a negligible effect on the energies but increase the costs during the CC iterations. The most accurate and recommeded variant is the CCSD(F12*) approximation [18], which gives essentially identical energies as CCSD(F12)-SP. Also available are the CCSD[F12] (Ref. [18]), CCSD-F12a (Ref. [165]) and CCSD-F12b (Ref. [166]) approximations as well as the perturbative corrections CCSD(2)F12 and CCSD(2*)F12 (see Refs. [18,167,168]). These approximations should only be used with ansatz 2 and the SP approach (i.e. fixed geminal amplitudes). Note that the CCSD-F12c method which is available in the molpro package is the same as CCSD(F12*). Since CCSD(F12*) is the original name that was introduced when the method was proposed for the first time in Ref. [18] users are asked to use in publications this abbreviation and cite the original reference [18].
For MP3 the approximations (F12*), and (F12) to a full F12 implementation become identical: they include all contributions linear in the coefficients cijkl. The explicitly-correlated MP4 method MP4(F12*) is defined as fourth-order approximation to CCSD(F12*)(T). Note that MP4(F12*) has to be used with the SP or fixed amplitude approach for the geminal coefficients cijkl. MP3(F12*) and MP4(F12*) are currently only available for closed-shell or unrestricted Hartree-Fock reference wavefunctions.
The CPU time for a CCSD(F12) calculation is approximately the sum of the CPU time for an MP2-F12 calculation with the same basis sets plus that of a conventional CCSD calculation multiplied by (1 + NCABS∕N), where N is the number of basis and NCABS the number of complementary auxiliary basis (CABS) functions (typically NCABS ≈ 2 - 3N). If the geminal coefficients are determined by solving Eq. (11.16) instead of using fixed amplitudes, the costs per CCSD(F12) iteration increase to ≈ (1 + 2NCABS∕N) times the costs for a conventional CCSD iteration. Irrespective how the geminal coefficients are determined, the disc space for CCSD(F12) calculations are approximatly a factor of ≈ (1 + 2NCABS∕N) larger than the disc space required for a conventional CCSD calculation. Note that this increase in the computational costs is by far outweighted by the enhanced basis set convergence.
In combination with the CCSD(F12*) approximation (and also CCSD[F12], CCSD-F12a, CCSD-F12b, CCSD(2)F12 and CCSD(2*)F12) the CPU time for the SP approach is only about 20% or less longer than for a conventional CCSD calculation within the same basis set.
CC calculations with restricted open-shell (ROHF) references: The MP2 and all CC calculations for ROHF reference wavefunctions are done by first transforming to a semi-canonical orbital basis which are defined by the eigenvectors of the occupied/occupied and virtual/virtual blocks of the Fock matrices of alpha and beta spin. No spin restrictions are applied in the cluster equations. This approach is sometimes also denoted as ROHF-UCCSD.
Note that if a frozen-core approximation is used, the semicanonical orbitals depend on whether the block-diagonalization of the Fock matrices is done in space of all orbitals or only in the space of the correlated valence orbitals. The two approaches lead thus to slightly different energies, but none of two is more valid or more accurate than the other. The ccsdf12, pnoccsd and ricc2 programs use the former scheme with the block-diagonalization done in the space of all molecular orbitals. The same scheme is used e.g. in the CFOUR program suite, but other codes as e.g. the implementation in MOLPRO use a block-diagonalization restricted to the active valence space.
Perturbative triples corrections: To achieve ground state energies with a high accuracy that systematically surpasses the acccuracy MP2 and DFT calculations for reaction and binding energies, the CCSD model has to be combined with a perturbative correction for connected triples. The recommended approach for the correction is the CCSD(T) model
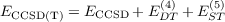 | (11.18) |
which includes the following two terms:
![(4) ∑ CCSD (2)
EDT = tμ2 ⟨μ2|[H, T3 ]|HF ⟩ (11.19)
∑μ2
ES(5T) = tCμC1SD⟨μ2|[H, T(32)]|HF ⟩ (11.20)
μ1](DOK182x.png)
![⟨ijk|[Hˆ, T2]|HF⟩
t(a2i)bjck = ------abc--------------- (11.21)
ϵa - ϵi + ϵb - ϵj + ϵc - ϵk](DOK183x.png)
Integral-direct implementation and resolution-of-the-identity approximation: The computationally most demanding (in terms floating point operations) steps of a CCSD calculation are related to two kinds of terms. One of the most costly steps is the contraction
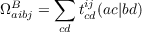 | (11.22) |
where a, b, c, and d are virtual orbitals. For small molecules with large basis sets or basis sets with
diffuse functions, where integral screening is not effective, it is the time-determing step and can
most efficiently be evaluated with a minimal operation count of  O2V 2 (where O and V are
number of, respectively occupied and virtual orbitals), if the 4-index integrals (ac|bd) in the MO
are precalculated and stored on file before the iterative solution of the coupled-cluster equation,
11.4 and 11.5, and the full permutational symmetries of tcdij, (ac|bd), and Ωaibj are
exploited. For larger systems, however, the storage and I/O of the integrals (ac|bd) leads to
bottlenecks. Alternatively, this contribution can be evaluated in an integral-direct way
as
O2V 2 (where O and V are
number of, respectively occupied and virtual orbitals), if the 4-index integrals (ac|bd) in the MO
are precalculated and stored on file before the iterative solution of the coupled-cluster equation,
11.4 and 11.5, and the full permutational symmetries of tcdij, (ac|bd), and Ωaibj are
exploited. For larger systems, however, the storage and I/O of the integrals (ac|bd) leads to
bottlenecks. Alternatively, this contribution can be evaluated in an integral-direct way
as
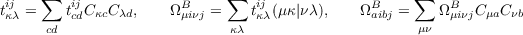 | (11.23) |
which, depending on the implementation and system, has formally a 2–3 times larger operation
count, but allows to avoid the storage and I/O bottlenecks by processing the 4-index integrals
on-the-fly without storing them. Furthermore, integral screening techniques can be
applied to reduce the operation count for large systems to an asymptotic scaling with
(4).
In TURBOMOLE only the latter algorithm is presently implemented. (For small systems other codes will therefore be faster.)
The other class of expensive contributions are so-called ring terms (in some publications denoted as C and D terms) which involve contractions of the doubles amplitudes taibj with several 4-index MO integrals with two occupied and two virtual indeces, partially evaluated with T1-dependent MO coefficients. For these terms the implementation in TURBOMOLE employs the resolution-of-the-identity (or density-fitting) approximation (with the cbas auxiliary basis set) to reduce the overhead from integral transformation steps. Due this approximation CCSD energies obtained with TURBOMOLe will deviate from those obtained with other coupled-cluster programs by a small RI error. This error is usually in the same order or smaller than the RI error for a RI-MP2 calculation for the same system and basis sets.
The RI approximation is also used to evaluate the 4-index integrals in the MO basis needed for the perturbative triples corrections.
Disc space requirements: In difference to CC2 and MP2, the CCSD model does no longer allow to avoid the storage of double excitation amplitudes (tabij) and intermediates of with a similar size. Thus, also the disc space requirements for CCSD calculations are larger than for RI-MP2 and RI-CC2 calculations for the same system. For a (closed-shell) CCSD ground state energy calculation the amount of disc space needed can be estimated roughly as
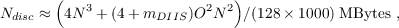 | (11.24) |
where N is the number of basis functions, O the number of occupied orbitals and mDIIS the number of vectors used in the DIIS procedure (by default 10, see Sec. 20.2.19 for details).
For (closed-shell) CCSD(T) calculations the required disc space is with
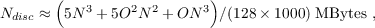 | (11.25) |
somewhat larger.
For calculations with an open-shell (UHF or ROHF) reference wavefunction the above estimates should be multiplied by factor of 4.
Memory requirements:
The CCSD and CCSD(T) implementation in Turbomole uses multi-pass algorithms to avoid
strictly the need to store arrays with a size of (N3) or (O2N2) or larger as complete array in
main memory. Therefore, the minimum memory requirements are relatively low—although it is
difficult to give accurate estimates for them.
On should, however, be aware that, if the amount of memory provided to the program in the data group $maxcor becomes too small compared to O2N2∕(128 × 1000) MBytes, loops will be broken in many small batches at the cost of increased I/O and a decrease in performance. As mentioned above, it is recommended to set $maxcor to 66–75% of the physical core memory available for the calculation.
Important options: The options to define the orbital and the auxiliary basis sets, the maximum amount of allocatable core memory ($maxcor), and the frozen-core approximation ($maxcor) have been mentioned above and described in the chapters on MP2 and CC2 calculations. Apart from this, CCSD and CCSD(T) calculations require very little additional input.
Relevant are in particular some options in the $ricc2 data group:
The options ccsd and ccsd(t) request, respectively, CCSD and CCSD(T) calculations. Since CCSD(T) requires the cluster amplitudes from a converged CCSD calculation, the option ccsd(t) also implies ccsd.
The number given for mxdiis defines the maximum number of vectors included in the DIIS procedure for the solution of the cluster equations. As mentioned above, it has some impact on the amount of disc space used by a CCSD calculation. Unless disc space becomes a bottleneck, it is not recommended to change the default value.
With maxiter one defines the maximum number of iterations for the solution of the cluster equations. If convergence is not reached within this limit, the calculation is stopped. Usually 25 iterations should be sufficient for convergence. Only in difficult cases with strong correlation effects more iterations are needed. It is recommended to increase this limit only if the reason for the strong correlation effects is known. (Since one reason could also be an input error as e.g. unreasonable geometries or orbital occupations or a wrong basis set assignment. With diffuse basis functions it is sometimes neccessary to tighten the integral screening threshold in $scftol.)
The two parameters conv and oconv define the convergence thresholds for the iterative solution of the cluster equations. Convergence is assumed if the change in the energy (with respect to the previous iteration) is smaller than 10-conv and the euclidian norm of the residual (the so-called vector function) smaller than 10-oconv. If conv is not given in the data group $ricc2 the threshold for changes in the energy is set to the value given in $denconv (by default 10-7). If oconv is not given in the data group $ricc2 the threshold for the residual norm is by default set to 10 ×conv. With the default settings for these thresholds, the energy will thus be converged until changes drop below 10-7 Hartree, which typically ensures an accuracy of about 1 μH. These setting are thus rather tight and conservative even for the calculation of highly accurate reaction energies. If for your application larger uncertainities for the energy are tolerable, it is recommended to use less tight thresholds, e.g. conv=6 or conv=5 for an accuracy of, respectively, at least 0.01 mH (0.03 kJ/mol) or 0.1 mH (0.3 kJ/mol). The settings for conv (and oconv) have not only an impact on the number of iterations for the solution of the cluster equations, but as they determine the thresholds for integral screening also on the costs for the individual iterations.
CCSD(T) energy with a second-order correction from the interference-corrected MP2-F12: The error introduced from a CCSD(T) calculation with a finite basis set can be corrected from second-order corrections of the the interference-corrected MP2-F12 (INT-MP2-F12) (Ref. [169]). The approximate CCSD(T)-INT-F12 at the basis set limit is given from
![∑
ECCSD(T)∕CBS ≈ ECCSD(T) + F ij[eMiPj2 -F12 - eMijP2].
i<j](DOK189x.png) | (11.26) |
From define, in the submenu $ricc2 select the ccsd(t) method and add the keyword intcorr
Then, switch on the f12 method (approximation A or B, inv or fixed). The corrected CCSD(T)-INT-F12 energy will be printed in the end of the calculation. It is highly recommended to start the CCSD(T)-INT-F12 calculation from a converged SCF calculation with symmetry, which is transfromed to C1. It is furthermore recommended to use Boys localized orbitals in the $rir12 submenu. A table with the corrected second-order pair-electron energies and the corresponding interference factors can also be printed in the output by using the keyword intcorr all instead of intcorr.
Excitation energies with CCSD: At the (conventional) CCSD level also electronic excitation energies can be computed. For this the data group $excitations has to be added (the same keyword as for CC2 applies). The implementation is currently restricted to vertical excitation energies (no transition moments or properties available) and in the closed–shell case to singlett excited states.
Note that for single-excitation dominated transitions CCSD is as CC2 correct through second-order and is not neccessarily more accurate than CC2. It is, however, for double excitations still correct through first-order, while CC2 describes double excitations only in a zero-order approximation. Therefore, CCSD results are more robust with respect to double excitation contributions to transitions and are thus usefull to check if CC2 is suitable for a certain problem.