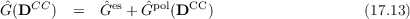
Polarizable embedding (PE) calculations are a based on a hybrid model of quantum mechanics and molecular mechanics (QM/MM) in which the classical region is represented by an electrostatic potential with up to octupole moments and induced point dipole moments. The main improvement over the more common QM/MM approaches without polarizable MM sites can be found for the description of electronic excitations but also for any other process which causes a significant change in the QM density and which is accompanied by a fast response of the environment.
In TURBOMOLE, only ground state energies computed with the dscf, ridft, and ricc2 module and electronic excitation properties based on RI-CC2 are implemented. The general theory is presented in ref. [208] and [209], the PERI-CC2 model and the TURBOMOLE implementation is described in ref. [210].
In the following, only the most important ideas are presented and discussed with a focus on the PERI-CC2 model. The essential concept is the introduction of an environment coupling operator Ĝ(DCC)
with the electrostatic contribution
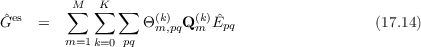
and the polarization contribution
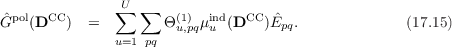
Here, Θm,pq(k) are multipole interaction integrals of order k and μuind are the induced dipoles which can be obtained from the electric field Fu and the polarizability αu at a site u:
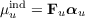 | (17.16) |
Because the induced dipoles depend on the electron density and vice versa, their computations enter the self-consistent part of the HF cycle. Introducing Ĝ(DCC) into standard equations for the HF reference state and the CC2 equations leads to a general PE-CC2 formulation. To maintain efficiency, a further approximation has been introduced which makes the operator only dependent on a CCS-like density term. These general ideas define the PERI-CC2 model and allow to formulate the corresponding Lagrangian expression
from which all PERI-CC2 equations including the linear response terms may be derived. Note that
the dependency on the density couples the CC amplitude and multiplier equations for the ground
state solution vector.
This coupling is avoided by the simplified polarizable embedding method (sPE) described in ref. [211].
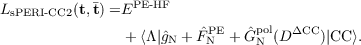 | (17.18) |
The subscript N indicates that the operator is normal ordered with respect to the Hartree–Fock state. Here, a polarization operator Ĝpol(D) was introduced,
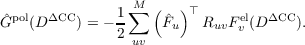 | (17.19) |
which depends on the elements of the difference density matrix DΔCC. These are defined as
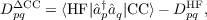 | (17.20) |
hence, they do not depend on the Lagrangian multipliers.
To carry out a PE-SCF calculation with the DSCF or RIDFT module, you have to specify the following in the control file:
An example for a polarizable embedding with coordinates given in Å, point charges and isotropic polarizabilities:
All values are given in atomic units (except coordinates if stated otherwise). These data are mandatory. An alternative input format can also be used by specifying daltoninp as option on the $point_charges line. The format is completely compatible with the current Dalton 2015 input format (The definition can be found in the manual from http://daltonprogram.org/www/documentation.html. See there for more information).
In addition, you can specify further options on the same line as the $point_charges flag. These are:
Limitations with respect to standard SCF computations:
The energy of a PE-SCF calculation printed in the output contains the following terms:
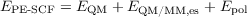 | (17.21) |
Here, EQM is the energy of the quantum mechanical method of your choice, EQM∕MM,es the electrostatic interaction energy between the QM and the MM region, and Epol the energy gain due to the total of induced dipole moments. If necessary, missing terms can be computed without knowledge of the electron distribution.
At the moment, TURBOMOLE does not offer the possibility to generate the necessary potentials or to create a potential file from a set of coordinates. Embedding potentials can be obtained from literature or generated by approaches like the LoProp method. [212] Atom centered polarizabilities are also available from other methods or from experiment. Finally, there are some polarizable force fields which, in principle, can be used for the PE method (for example, the AMOEBA force field).
Apart from the definition of the embedding described above, the input for PERI-CC2 calculations is the same as without polarizable embedding.
There are several limitations for the use of PERI-CC2:
![¯ ˆ ˆ ˆ 1 ˆ2
LPERI-CC2(t,t) = EPE-HF + ⟨HF |W (T1 + T2 + 2T1 )|HF⟩ +
∑ ¯t ⟨μ | ˜W + [ˆFPE,Tˆ]+ [ ˜W ,T ˆ]|HF⟩+
μ1 μ1 1 1 2
∑ PE
¯tμ2⟨μ2| ˜W + [ˆF ,Tˆ2]|HF ⟩
μ2
- 1∑ F elec(D Δ′)RuvF elec(D Δ′) (17.17)
2 uv u v](DOK289x.png)