 ⋅ KIJ ⋅
⋅ KIJ ⋅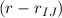 2
2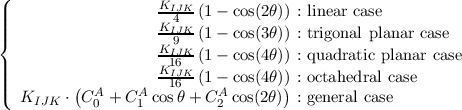
 ⋅ V ϕ ⋅
⋅ V ϕ ⋅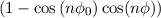
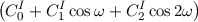
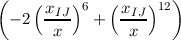
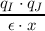
uff preoptimizes a structure and calculates an analytical Hessian which can be used as a start Hessian in a geometry optimization. This will accelerate the convergence of an optimizations. For optimizations in cartesian space this will be faster by a factor of two for any molecule.
You have to generate cartesian coordinates (file coord), nothing else. You can start an single-point calculation calculation by typing
uff
To start an uff geometry optimization, one has to change the number of cycles (parameter maxcycle) in the block $uff in the file control. The ouput is the optimized structure (file coord), the analytical gradient (file uffgradient) and the analytical cartesian hessian (file uffhessian0-0). Furthermore the control file will be modified:
These commands have the effect to inititialize the force constant matric for a geometry optimization with the hessian one.
In some cases uff cannot recognize the connectivity, then one can specify the connectivity in the file ufftopology. The program will calculate the bond, angle, torsion, inverison and non-bonded terms (force field terms) based on the connectivity specified in the topology file.
The uff implementation follows the paper by Rappé [7]. The energy expression in uff is
as follows:
| EUFF = | ∑
NB
⋅ KIJ ⋅2 | (5.1) | |
| + | ∑
NA
| ||
| + | ∑
NT
⋅ V ϕ ⋅ | ||
| + | ∑
NI
V ω ⋅ | ||
| + | ∑
Nnb
DIJ ⋅ | ||
| + | ∑
Nnb
|
| C2A | = 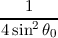 | (5.2) |
| C1A | = -4 ⋅ C 2A cosθ 0 | (5.3) |
| C0A | = C
2A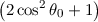 | (5.4) |
the numbers of the bond-, angle-, torsion-, inversion- and the non bonded-terms.
forceconstants of the bond- and angle-terms.
bond distance and natural bond distance of the two atoms I and J.
angle and natural angle for three atoms I - J - K.
Fourier coefficients of the general angle terms.
torsion angle and natural torison angle of the atoms I -J - K - L.
height of the torsion barrier.
periodicity of the torsion potential.
inversion- or out-of-plane-angle at atom I.
height of the inversion barrier.
Fourier coefficients of the inversions terms.
distance and natural distance of two non bonded atoms I and J.
depth of the Lennard–Jones potential.
partial charge of atoms I and dielectric constant.
One major difference in this implementation concerns the atom types. The atom types in Rappé’s paper have an underscore "_". In the present implementation an sp3 C atom has the name "C 3" instead of "C_3". Particularly the bond terms are described with the harmonic potential and the non-bonded van der Waals terms with the Lennard–Jones potential. The partial charges needed for electrostatic nonbond terms are calculated with the Charge Equilibration Modell (QEq) from Rappé [43]. There is no cutoff for the non-bonded terms.
The relaxation procedure distinguishes between molecules wih more than 90 atoms and molecules with less atoms. For small molecules it consists of a Newton step followed by a linesearch step. For big molecules a quasi-Newton relaxation is done. The BFGS update of the force-constant matric is done [38,44–46]. Pulay’s DIIS procedure is implemented for big molecule to accelarate the optimization [37,47].
The coordinates for any single atom can be fixed by placing an ’f’ in the third to eighth column of the chemical symbol/flag group. As an example, the following coordinates specify acetone with a fixed carbonyl group: