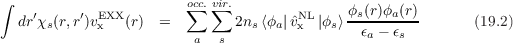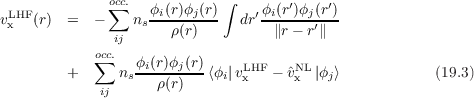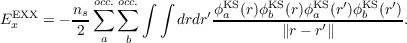
Approximations to the exchange-correlation (XC) functional of the Kohn-Sham (KS) Density Functional Theory (DFT) can be classified by the so-called “Jacob’s ladder.” The ground on which the ladder lies is the Hartree approximation (XC energy is zero), and the first rung is the local density approximation (LDA) in which the XC energy density is a simple local function of the density. The second rung of the Jacob’s ladder is the generalized gradient approximation (GGA): in this case the XC energy density depends also on the gradient of the density. In the third rung (meta-GGA) an additional variable is used, the Kohn-Sham kinetic energy density which allows, e.g., to construct self-correlation-free functionals. Functionals in the above rungs can have high accuracy for different class of problems in chemistry and solid-state physics, but their main limitation is the self-interaction error (SIE) [219–222]. To avoid the SIE the exchange must be treated exactly and this can be achieved by functionals in the fourth rung which depend explicitly on all the occupied KS orbitals. In the KS formalism the EXX (exact-exchange) energy is (for closed-shell systems ns = 2) [219–222]:
|
| (19.1) |
i.e. the same functional form of the Hartree-Fock (HF) exchange but computed with KS orbitals
which are obtained using a self-consistent local EXX potential. At this point we should recall that
hybrid DFT functionals (including HF exchange), doesn’t belong to the KS formalism: in hybrid
DFT, in fact, the non-local HF exchange operator  xNL(r,r′) = -∑
aocc.
xNL(r,r′) = -∑
aocc.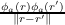 is
employed in the self-consistent (Generalized Kohn-Sham) equations determining the
orbitals.
is
employed in the self-consistent (Generalized Kohn-Sham) equations determining the
orbitals.
While LDA, GGA, meta-GGA and hybrid functionals are implemented (for ground-state calculations) in the dscf and ridft, the odft module considers functionals of the fourth rung. Currently exchange-only orbital-dependent approaches are implemented in the odft module. The EXX KS local potential (vxEXX(r)) can be obtained using the optimized effective potential (OEP) method (in each self-consistent step): [220–223]:
where χs(r,r′) = ∑ aocc.2ns ∑ svir.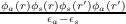 is the non-interacting density
response.
is the non-interacting density
response.
An effective approximation to the OEP-EXX potential is given by the Localized Hartree–Fock (LHF) potential [219] which is given by
where the first term is called Slater potential and the second term correction term. If terms i≠j are neglected in the correction term, the Krieger-Li-Iafrate (KLI) potential [224] is obtained. Note that the Eq. (19.3) depends only on occupied orbitals, whereas Eq. (19.2) depends also on virtual orbitals. The LHF total energy is assumed to be the EXX total energy, even if LHF is not variational (although the deviation from the EXX energy is very small, usually below 0.01%). The LHF potential is equivalent to the Common Energy Denominator Approximation (CEDA) [225] and to the Effective Local Potential (ELP) [226].Both OEP-EXX and LHF (in contrast to functionals of the first three rungs) satisfy the HOMO condition [224]
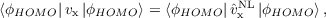 | (19.4) |
and the asymptotic relation [227,228]
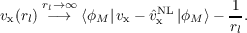 | (19.5) |
where ϕM is the highest occupied orbital which do not have a nodal surface in the asymptotic region along direction rl. Considering together with condition (19.4), we finally obtain that:
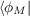 vx -
vx - xNL
xNL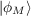 - 1∕r.
- 1∕r.Both OEP-EXX and LHF gives total energies very close to the Hartree-Fock one (actually ELHF > EEXX > EHF), thus, without an appropriate correlation functional, these methods are not suitable for thermochemistry. On the other hand OEP-EXX and LHF give very good KS orbital spectra. In fact the eigenvalues of the HOMO is very close to the Hartree-Fock and to exact ionization potential (I.P): this is in contrast to functional of the first three rungs which underestimate the HOMO energy by several eVs. In addition a continuum set of bound unoccupied orbitals are obtained. Thus OEP-EXX or LHF KS orbitals are very good input quantities for computing NMR shielding constants [229], energy-levels in hybrid interfaces [230] and TD-DFT excitation energies [231] (the latter using LDA/GGA kernels, not the hybrid ones).